Bioinformatic analysis#
1. Preparing data for processing#
1.1 Unzipping data#
The sequencing data for this project came as a single zipped file (HKeDNAworkshop2023.zip) which we moved to the sequenceData/2-raw subdirectory. To check if our sequence data is in the appropriate subfolder, let us use the ls -ltr command again.
ls -ltr sequenceData/2-raw
Output
-rw-r--r-- 1 ednaw01 others 568074346 Oct 21 10:59 HKeDNAworkshop2023.zip
Warning
The sequencing data has the extension .zip. This is not the same as the extension .gz, which is a g-zipped file structure frequently provided by the sequencing service during file transfer. The following set of commands that deal with the .zip extension are specific for this data type, a result due to the alteration of files after subsetting from the original data. It is unlikely that these steps will be necessary for your project data if your data was received from a sequencing service.
Since the data comes zipped, we need to use the unzip command to get access to the sequencing files. We can use the d parameter to tell unzip where we would like to place the unzipped files. In our tutorial, we will place the files in a subdirectory of sequenceData/2-raw/ called sequenceData/2-raw/unzipped/. Note that we do not need to create this subdirectory first. The unzip command will generate this subdirectory automatically if it not yet exists.
unzip sequenceData/2-raw/HKeDNAworkshop2023.zip -d sequenceData/2-raw/unzipped/
Output
Archive: sequenceData/2-raw/HKeDNAworkshop2023.zip
inflating: sequenceData/2-raw/unzipped/COI_100_HK37_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK37_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK38_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK38_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK39_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK39_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK40_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK40_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK41_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK41_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK42_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK42_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK49_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK49_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK50_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK50_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK51_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK51_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK52_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK52_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK53_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK53_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK54_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_100_HK54_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK37_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK37_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK38_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK38_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK39_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK39_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK40_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK40_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK41_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK41_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK42_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK42_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK49_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK49_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK50_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK50_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK51_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK51_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK52_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK52_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK53_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK53_2.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK54_1.fastq
inflating: sequenceData/2-raw/unzipped/COI_500_HK54_2.fastq
inflating: sequenceData/2-raw/unzipped/NTC_1_1.fastq
inflating: sequenceData/2-raw/unzipped/NTC_1_2.fastq
inflating: sequenceData/2-raw/unzipped/NTC_2_1.fastq
inflating: sequenceData/2-raw/unzipped/NTC_2_2.fastq
inflating: sequenceData/2-raw/unzipped/NTC_3_1.fastq
inflating: sequenceData/2-raw/unzipped/NTC_3_2.fastq
Unzipping the HKeDNAworkshop2023.zip file has generated a bunch of files for all of the samples in our experiment, each represented with a forward (_1.fastq) and reverse (_2.fastq) fastq file.
1.1.1 Counting files (pipe |)#
Now that we are getting more familiar with the Terminal and bash commands, let’s introduce something that is called a pipe, which is represented by the | symbol. A pipe or placing the | in your terminal allows you to use the output from one command as the input of a second command. Hence, the terminology “pipe”, as | acts as a connection between two different commands. To show you the power of piping commands together, let’s look at the exercise of how you might count the number of samples you have in our data set.
STEP 1: To solve the issue of counting the number of samples, we can use a step-by-step approach. First, we can use a command you already know, the ls -1 command to list all the files in the directory. The -1 parameter will print the files into a single column. Let’s see the output of this command first before we continue.
ls -1 sequenceData/2-raw/unzipped/
Output
COI_100_HK37_1.fastq
COI_100_HK37_2.fastq
COI_100_HK38_1.fastq
COI_100_HK38_2.fastq
COI_100_HK39_1.fastq
COI_100_HK39_2.fastq
COI_100_HK40_1.fastq
COI_100_HK40_2.fastq
COI_100_HK41_1.fastq
COI_100_HK41_2.fastq
COI_100_HK42_1.fastq
COI_100_HK42_2.fastq
COI_100_HK49_1.fastq
COI_100_HK49_2.fastq
COI_100_HK50_1.fastq
COI_100_HK50_2.fastq
COI_100_HK51_1.fastq
COI_100_HK51_2.fastq
COI_100_HK52_1.fastq
COI_100_HK52_2.fastq
COI_100_HK53_1.fastq
COI_100_HK53_2.fastq
COI_100_HK54_1.fastq
COI_100_HK54_2.fastq
COI_500_HK37_1.fastq
COI_500_HK37_2.fastq
COI_500_HK38_1.fastq
COI_500_HK38_2.fastq
COI_500_HK39_1.fastq
COI_500_HK39_2.fastq
COI_500_HK40_1.fastq
COI_500_HK40_2.fastq
COI_500_HK41_1.fastq
COI_500_HK41_2.fastq
COI_500_HK42_1.fastq
COI_500_HK42_2.fastq
COI_500_HK49_1.fastq
COI_500_HK49_2.fastq
COI_500_HK50_1.fastq
COI_500_HK50_2.fastq
COI_500_HK51_1.fastq
COI_500_HK51_2.fastq
COI_500_HK52_1.fastq
COI_500_HK52_2.fastq
COI_500_HK53_1.fastq
COI_500_HK53_2.fastq
COI_500_HK54_1.fastq
COI_500_HK54_2.fastq
NTC_1_1.fastq
NTC_1_2.fastq
NTC_2_1.fastq
NTC_2_2.fastq
NTC_3_1.fastq
NTC_3_2.fastq
STEP 2: Once we have a list, we can use a count function such as wc which stands for word count. However, we want to count lines, not words, so we need to add the -l parameter to specify this. When we pipe these two commands together, the Terminal window will output the number of files in our folder. Let’s try!
ls -1 sequenceData/2-raw/unzipped/ | wc -l
Output
54
STEP 3: Remember that we wanted to know the number of samples for which we have data files and that each sample was comprised of one forward (_1.fastq) and one reverse (_2.fastq) file. So, for the last step, we need to divide the number of files by 2. The easiest way to accomplis this step is to pipe the number of files in our directory to an AWK command (a separate language in the Terminal) that takes the number and divides it by 2. For this last command, we specify that we would like to print the first item ($1), which in our case is the number of files, and divide it by two (/2).
ls -1 sequenceData/2-raw/unzipped | wc -l | awk '{print $1/2}'
Output
27
By using one line of code and two pipes, we have shown that we have 27 samples in our data set. In our tutorial data set, these 27 samples are comprised of 3 negative controls, and 2 size fractions * 3 replicates * 4 sites.
Tip
AWK, sed, and grep are very powerful computer languages to process text. Throughout this tutorial, we will be using snippets of code from these three computer languages. Unfortunately, the syntax of these languages is quite complex in my opinion. I recommend you to investigate these languages on your own time if bioinformatics is of interest to you. However, to keep this tutorial beginner friendly, we will not expand on the syntax further.
1.2 Fastq and Fasta file structure#
When unzipping the HKeDNAworkshop2023.zip file, we have revealed our sequence data files. Those sequence data files have the extension .fastq. Sequence data is most frequently represented as either .fastq or .fasta files. Both are simple text files that are structured in a particular way.
For example, within .fastq files each sequence record is represented by 4 lines. The first line contains the header information and the line starts with the symbol @. The second line contains the actual sequence. The third line can contain metadata, but usually is restricted to the + symbol. The fourth line provides the quality of the base call and should, therefore, have the same length as the second line containing the sequence. We can inspect the file structure of one of our sequence files we just unzipped using the head or tail command. Both commands print the first or last N number of lines in a document, respectively. By using the -n parameter, we can specify the number of lines to be printed.
head -n 4 sequenceData/2-raw/unzipped/COI_100_HK37_1.fastq
Output
@M04617:136:000000000-J6LL9:1:1101:14901:1936 1:N:0:31
GGTACTGGATGAACAGTATATCCCCCCCTAAGCTCCAATATTGCCCACGCCGGGGCGTCTGTTGACCTTGCTATCTTTAGGCTACACTTGGCTGGGGTTTCTTCTCTACTCGGGGCTGTAAACTTTATTAGAACTATCGCTAACCTGCGAGCTTTAGGGCTAATTCTTGACCGTATAACACTATTCACATGATCAGTTCTTATCACCGCCATCCTTCTCCTTCTTTCTCTACCTGTTCTCGCAGGGGCTAT
+
3AA@AFFFCFFFBG5GGBGBFGHCFE?AEA233FFE33D3FGBFFGBA0AEE???E?EEEAGHFB@BGGBEGDFCGGG43F3GBGBFFGFBGFECCCFDGDGHFHFHB>GFA///AD?G1<1<FGHBGH1DB1>FBGD..C../<GCF---;AC0000.:C:0CBBFFFBBB-C.C/0B0:F/FFB099C9BCF00;C0FF/BB/B..---;/.BFBFBBFFFFFFFFFF/BFFBFFFFF.9-.---9@.B
.fasta files, on the other hand, are simple text files structured in a slightly different way, whereby each sequence record is represented by 2 lines. The first line contains the header information and the line starts with the symbol >. The second line contains the actual sequence. The third and fourth line that are in the .fastq files are missing in the .fasta files, as this file structure does not incorporate the quality of a sequence. We can inspect the file structure of a .fasta file by using the head or tail command on the reference database.
tail -n 2 sequenceData/7-refdb/newDBcrabsCOIsintax.fasta
Output
>AJ488637;tax=d:Eukaryota,p:Mollusca,c:Gastropoda,o:Littorinimorpha,f:Littorinidae,g:Littoraria,s:Littoraria_scabra
TCTTGCAGGCAACCTGGCTCACGCCGGGGGCTCTGTAGATCTAGCAATTTTTTCACTCCATCTAGCCGGTGTGTCTTCTATTTTAGGGGCTGTAAATTTCATTACAACCATCATTAATATGCGATGACGAGGTATGCAGTTTGAACGTCTACCTCTCTTTGTTTGATCAGTAAAGATTACAGCTATTCTTCTTCTTTTATCTCTCCCAGTTTTAGCTGGTGCAATTACCATACTCTTAACGGATCGAAACTTCAATACTGCCTTCTTTGACCCTGCCGGAGGAGGAGATCC
Exercise 2
Knowing the file structure of the .fastq and .fasta sequence files. How would you calculate the number of sequences incorporated in the files sequenceData/2-raw/unzipped/COI_100_HK37_1.fastq and sequenceData/7-refdb/leray_COI_sintax.fasta?
Answer 2
grep -c "^@M04617" sequenceData/2-raw/unzipped/COI_100_HK37_1.fastq
grep -c "^>" sequenceData/7-refdb/leray_COI_sintax.fasta
wc -l sequenceData/2-raw/unzipped/COI_100_HK37_1.fastq
wc -l sequenceData/7-refdb/leray_COI_sintax.fasta
with open('sequenceData/2-raw/unzipped/COI_100_HK37_1.fastq', 'r') as infile:
x = len(infile.readlines())
print('Total lines:', x)
1.3 Changing file names#
When we inspect the file names of our .fastq files by using the ls command, we can see that they contain the _ symbols.
Fig. 4 : A list of the fastq file names#
While _ symbols will not cause downstream incompatability issues, some symbols in file names can cause problems downstream with certain programs. One frequently observed symbol that affects one of the programs we will use during our bioinformatic pipeline is the dash - symbol. While not pertinent for our tutorial file names, it is important to know how to remove or change such symbols in file names in the Terminal without having do to this manually. As an exercise, let’s replace all the underscores _ to dashes - and back to show you how the code would work if you need it in the future.
Luckily there is a simple perl one-liner to batch replace characters in file names using the rename command. We can tell rename that we want to substitute a pattern by providing the s parameter. The /_/-/ parameter tells rename the pattern we want to substitute, i.e., replace _ with -. Finally, we tell rename that we want to replace all instances using the g or global parameter. Without this parameter, rename would only substitute the first instance. The * at the end of the code tells the computer to go over all the files in the unzipped/ directory.
cd sequenceData/2-raw/
rename 's/_/-/g' unzipped/*
Fig. 5 : A list of fastq file names whereby the underscores are replaced by dashes#
rename 's/-/_/g' unzipped/*
cd ../../
Fig. 6 : A list of fastq file names whereby the dashes are replaced back by underscores#
1.4 Quality control of raw data#
Up to this point, we have unzipped our sequence data, checked the file structure, and batch renamed our files to exclude problematic symbols in the file names. Before processing the contents of each file, it is best to check the quality of the raw sequence data to ensure no problems were faced during sequencing. The programs we will be using to check the quality of all of our files are FASTQC and MULTIQC. First, we will generate reports for all of the files using FastQC. We can specify the output folder using the -o parameter and the number of threads or cores using the -t parameter.
Warning
Make sure to change the value of the -t parameter to the number of cores available on your system!
To run FASTQC on the supercomputer, we can modify the template script sequenceData/1-scripts/example-script.sh. We can open text documents with CLI text editors, such as nano.
nano sequenceData/1-scripts/example-script.sh
When opening the script, we can add the following line of code to execute the FASTQC command.
fastqc sequenceData/2-raw/unzipped/* -o sequenceData/3-fastqc/ -t 8
We can exit out of the editor by pressing ctrl + x and y. We will change the name of the script to fastqc_raw.sh and press y to save under a different name. After completing these steps, you should end back up in the normal Terminal window. To check if we created this new script, let’s use the ls -ltr command again.
ls -ltr sequenceData/1-scripts
Output
-rw-r--r-- 1 ednaw01 others 776 Oct 21 10:59 example-script.sh
-rw-r--r-- 1 ednaw01 others 169 Oct 21 10:59 module_load
-rw-r--r-- 1 ednaw01 others 833 Oct 21 11:33 fastqc_raw.sh
To run the command on the supercomputer, we can use the sbatch command.
sbatch sequenceData/1-scripts/fastqc_raw.sh
Once this command is executed, it will be placed in the queue to be run on the cluster. We can use the squeue command to check if our code is running or if the job is finished. It is best to specify your jobs specifically using the -u or user parameter, followed by your user name.
squeue -u ednaw01
Warning
Make sure to change the -u parameter to your user name!
Output
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
Once finished, FastQC will generate a .html report for every single file in the subdirectory sequenceData/2-raw/unzipped/. Let’s open one report to see what it contains. The .html reports are stored in the subdirectory sequenceData/3-fastqc/. From the FastQC .html reports, we are particularly interested in the summary tab, as well as the per base sequence quality and sequence length distribution figures.
Since we are working on the supercomputer, we need to download the .html report to our computer before we can open it. To do this, let’s open a new Terminal window that is not connected to the supercomputer, move to the Desktop directory using the cd command and download the file using the scp command, which stands for secure copy. While not essential in this case, you can add the -r parameter to allow the download of directories. We also need to specify the server location of the file to download and where we would like to download the file to on our system.
cd Desktop
scp -r ednaw01@hpc2021-io1.hku.hk:~/ednaw01/sequenceData/3-fastqc/COI_100_HK37_1_fastqc.html ./
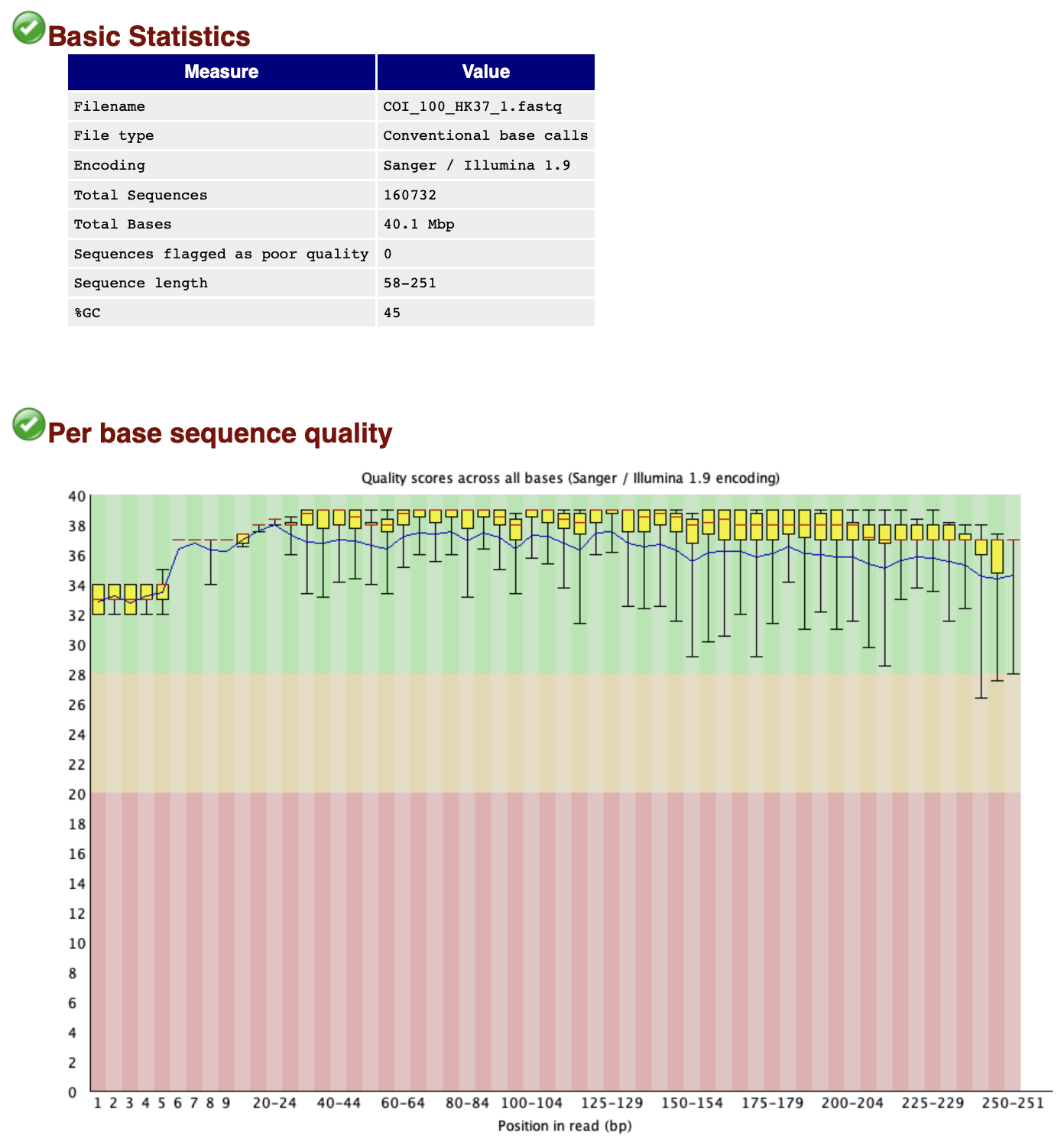
Fig. 7 : A FastQC report of the raw sequencing data.#
The quality of file COI_100_HK37_1.fastq is looking very good and the number of sequences reported for this sample (160,732 reads) is a good sequencing depth as well!
Opening each report for the 54 files separately, however, is quite tedious and makes it difficult to compare differences between files. Luckily, multiQC was developed to collate these reports into a single .html report. We can use the . symbol in combination with the path to specify a minimal output to the Terminal window. The -o parameter let’s us set the output directory.
Again, to run this on the supercomputer, we need to modify the initial template script and add in the multiqc code.
nano sequenceData/1-scripts/example-script.sh
multiqc sequenceData/3-fastqc/. -o sequenceData/3-fastqc/
Press ctrl + x and y to save the file and change the name to multiqc_raw.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/multiqc_raw.sh

Fig. 8 : Terminal output for multiQC#
The multiQC program will combine all 54 FastQC reports into a single .html document. Let’s download and open this file to see how our raw sequence data is looking like.
scp -r ednaw01@hpc2021-io1.hku.hk:~/ednaw01/sequenceData/3-fastqc/multiqc_report.html ./
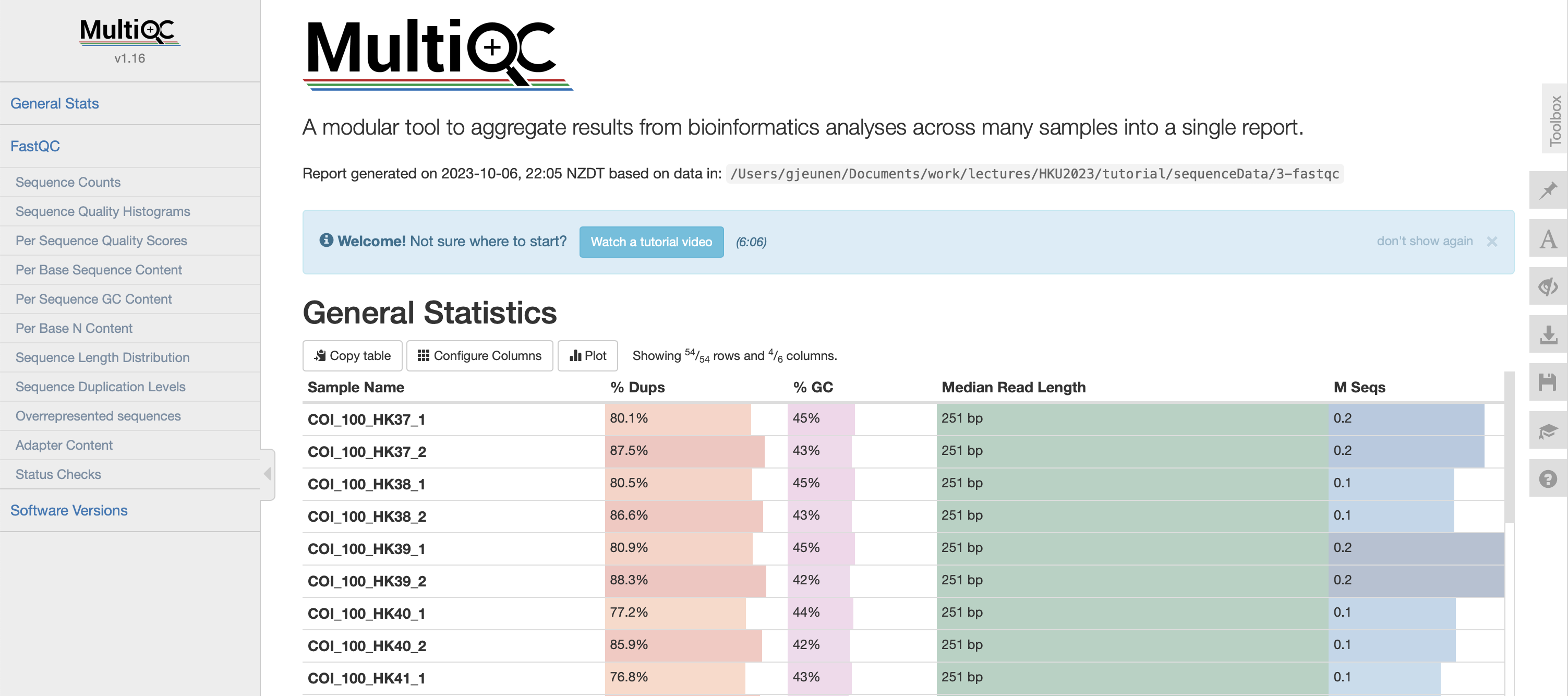
Fig. 9 : The .html multiQC report#
The sequence quality, average read length, and number of sequences are all very comparable between the different files representing the samples. This is an excellent starting point! Interestingly, note the difference in length, read depth, and quality with the control samples. As those are our negative controls, these differences aren’t worrysome. If, on the other hand, some samples were looking like this, you might want to consider resequencing those samples to achieve a similar sequencing depth.
2. Merging forward and reverse reads#
2.1 A single sample example#
Once we have assessed the quality of the raw sequence data and did not observe any issues, we can move ahead with merging the forward and reverse reads of each sample. As a reminder, we have 54 .fastq files, indicating that we have 27 samples in our experiment, one forward and one reverse file for each sample.
For this experiment and particular library, we have amplified a ~313 bp fragment of the COI gene (excluding primer-binding regions). With the sequencing run specifications of a MiSeq 2x250 paired-end V2 sequencing kit, we have a partial overlap between the forward and reverse reads in the middle of the amplicon region. This overlap is essential for successful merging. Today, we will be using the --fastq_mergepairs command in VSEARCH to merge reads between the forward and reverse .fastq sequence files. We can specify the reverse sequence file through the --reverse parameter and the output file through the --fastqout parameter. To check the options available within the program, you can always read the documentation and pull up the help vignette by executing vsearch --help in the Terminal window. Before merging reads immediately on all samples, it is best to play around with the parameters on a single sample.
nano sequenceData/1-scripts/example-script.sh
vsearch --fastq_mergepairs sequenceData/2-raw/unzipped/COI_100_HK37_1.fastq --reverse sequenceData/2-raw/unzipped/COI_100_HK37_2.fastq --fastqout sequenceData/2-raw/COI_100_HK37merged.fastq
Press ctrl + x and y to save the file and change the name to merge_single.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/merge_single.sh
Output
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
160732 Pairs
146040 Merged (90.9%)
14692 Not merged (9.1%)
Pairs that failed merging due to various reasons:
144 too few kmers found on same diagonal
5751 too many differences
8584 alignment score too low, or score drop too high
213 staggered read pairs
Statistics of all reads:
249.48 Mean read length
Statistics of merged reads:
359.43 Mean fragment length
27.01 Standard deviation of fragment length
0.32 Mean expected error in forward sequences
0.53 Mean expected error in reverse sequences
0.24 Mean expected error in merged sequences
0.22 Mean observed errors in merged region of forward sequences
0.56 Mean observed errors in merged region of reverse sequences
0.77 Mean observed errors in merged region
Using the default settings, we managed to merge 90.9% of reads. For this tutorial, we’ll take that as sufficient. However, for your own project, you might want to explore the various options within the fastq_mergepairs command further to try and increase the percentage of reads merged. From the vsearch --fastq_mergepairs output that was printed to the Terminal window, we can see that VSEARCH reports the number of observed pairs in the raw data files, the number of reads successfully merged, and the number of reads unable to be merged. Additionally, information on why reads couldn’t be merged and statistics about merged reads are provided.
2.2 Batch merging reads#
Since we need to merge sequences for 27 samples, we will introduce a new coding concept, the for loop. A for loop allows us to move through a list and execute a command on each item. While we will not go into too much detail for this tutorial, the basic syntax of for loops is as follows:
for file in list
do
execute command
done
Basically, the first line tells us that the program will go through a list and for each item in the list, do something (line 2), which in this case is execute command (line 3). Once the program has executed the command in all items within the list, we need to close the loop by specifying done (line 4). So, with a for loop, we can tell the computer to merge reads for our 27 samples automatically, saving us the time to have to execute the code to merge reads 27 times by ourselves.
Within our for loop to merge reads for all samples, we will generate a list of all forward sequencing files (*_1.fastq). Before writing the code to merge reads in our for loop, we will first print which sample is being merged, as the VSEARCH output does not specify this (echo "Merging reads for: ${R1/_1.fastq/}"). To print to the Terminal, we can use the echo command. With the last parameter for the echo command (${R1/_1.fastq/}"), we specify that from our forward sequencing file name $R1, substitute _1.fastq with nothing. After the echo command, we execute the vsearch --fastq_mergepairs command as we did above, but we need to use the for loop syntax to tell the program what our forward, reverse, and output file names are. Remember that those names change when the program iterates over the list, so we cannot hard code them into the command.
rm sequenceData/2-raw/COI_100_HK37merged.fastq
nano sequenceData/1-scripts/example-script.sh
cd sequenceData/2-raw/unzipped/
for R1 in *_1.fastq
do
echo "\n\nMerging reads for: ${R1/_1.fastq/}"
vsearch --fastq_mergepairs ${R1} --reverse ${R1/_1.fastq/_2.fastq} --fastqout ../${R1/1.fastq/merged.fastq}
done
Press ctrl + x and y to save the file and change the name to merge_batch.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/merge_batch.sh
Output
Merging reads for: COI_100_HK37
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
160732 Pairs
146040 Merged (90.9%)
14692 Not merged (9.1%)
Pairs that failed merging due to various reasons:
144 too few kmers found on same diagonal
5751 too many differences
8584 alignment score too low, or score drop too high
213 staggered read pairs
Statistics of all reads:
249.48 Mean read length
Statistics of merged reads:
359.43 Mean fragment length
27.01 Standard deviation of fragment length
0.32 Mean expected error in forward sequences
0.53 Mean expected error in reverse sequences
0.24 Mean expected error in merged sequences
0.22 Mean observed errors in merged region of forward sequences
0.56 Mean observed errors in merged region of reverse sequences
0.77 Mean observed errors in merged region
Merging reads for: COI_100_HK38
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
129473 Pairs
118917 Merged (91.8%)
10556 Not merged (8.2%)
Pairs that failed merging due to various reasons:
116 too few kmers found on same diagonal
4920 too many differences
5434 alignment score too low, or score drop too high
86 staggered read pairs
Statistics of all reads:
249.92 Mean read length
Statistics of merged reads:
361.90 Mean fragment length
20.07 Standard deviation of fragment length
0.30 Mean expected error in forward sequences
0.54 Mean expected error in reverse sequences
0.25 Mean expected error in merged sequences
0.21 Mean observed errors in merged region of forward sequences
0.58 Mean observed errors in merged region of reverse sequences
0.79 Mean observed errors in merged region
Merging reads for: COI_100_HK39
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
181198 Pairs
162326 Merged (89.6%)
18872 Not merged (10.4%)
Pairs that failed merging due to various reasons:
137 too few kmers found on same diagonal
2 multiple potential alignments
6778 too many differences
11862 alignment score too low, or score drop too high
1 overlap too short
92 staggered read pairs
Statistics of all reads:
250.08 Mean read length
Statistics of merged reads:
362.18 Mean fragment length
18.32 Standard deviation of fragment length
0.33 Mean expected error in forward sequences
0.48 Mean expected error in reverse sequences
0.24 Mean expected error in merged sequences
0.23 Mean observed errors in merged region of forward sequences
0.50 Mean observed errors in merged region of reverse sequences
0.73 Mean observed errors in merged region
Merging reads for: COI_100_HK40
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
130928 Pairs
115722 Merged (88.4%)
15206 Not merged (11.6%)
Pairs that failed merging due to various reasons:
144 too few kmers found on same diagonal
4604 too many differences
10378 alignment score too low, or score drop too high
80 staggered read pairs
Statistics of all reads:
250.12 Mean read length
Statistics of merged reads:
362.11 Mean fragment length
19.02 Standard deviation of fragment length
0.30 Mean expected error in forward sequences
0.49 Mean expected error in reverse sequences
0.23 Mean expected error in merged sequences
0.23 Mean observed errors in merged region of forward sequences
0.52 Mean observed errors in merged region of reverse sequences
0.75 Mean observed errors in merged region
Merging reads for: COI_100_HK41
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
115679 Pairs
102320 Merged (88.5%)
13359 Not merged (11.5%)
Pairs that failed merging due to various reasons:
108 too few kmers found on same diagonal
2 multiple potential alignments
4047 too many differences
9067 alignment score too low, or score drop too high
135 staggered read pairs
Statistics of all reads:
250.07 Mean read length
Statistics of merged reads:
361.47 Mean fragment length
20.81 Standard deviation of fragment length
0.28 Mean expected error in forward sequences
0.54 Mean expected error in reverse sequences
0.23 Mean expected error in merged sequences
0.20 Mean observed errors in merged region of forward sequences
0.59 Mean observed errors in merged region of reverse sequences
0.79 Mean observed errors in merged region
Merging reads for: COI_100_HK42
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
164258 Pairs
148463 Merged (90.4%)
15795 Not merged (9.6%)
Pairs that failed merging due to various reasons:
112 too few kmers found on same diagonal
1 multiple potential alignments
5409 too many differences
10244 alignment score too low, or score drop too high
29 staggered read pairs
Statistics of all reads:
250.39 Mean read length
Statistics of merged reads:
363.83 Mean fragment length
11.18 Standard deviation of fragment length
0.29 Mean expected error in forward sequences
0.48 Mean expected error in reverse sequences
0.23 Mean expected error in merged sequences
0.21 Mean observed errors in merged region of forward sequences
0.51 Mean observed errors in merged region of reverse sequences
0.72 Mean observed errors in merged region
Merging reads for: COI_100_HK49
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
97006 Pairs
91171 Merged (94.0%)
5835 Not merged (6.0%)
Pairs that failed merging due to various reasons:
51 too few kmers found on same diagonal
2739 too many differences
3039 alignment score too low, or score drop too high
6 staggered read pairs
Statistics of all reads:
250.31 Mean read length
Statistics of merged reads:
364.45 Mean fragment length
10.51 Standard deviation of fragment length
0.24 Mean expected error in forward sequences
0.49 Mean expected error in reverse sequences
0.23 Mean expected error in merged sequences
0.18 Mean observed errors in merged region of forward sequences
0.54 Mean observed errors in merged region of reverse sequences
0.72 Mean observed errors in merged region
Merging reads for: COI_100_HK50
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
85963 Pairs
80114 Merged (93.2%)
5849 Not merged (6.8%)
Pairs that failed merging due to various reasons:
48 too few kmers found on same diagonal
2669 too many differences
3127 alignment score too low, or score drop too high
5 staggered read pairs
Statistics of all reads:
250.46 Mean read length
Statistics of merged reads:
364.77 Mean fragment length
6.27 Standard deviation of fragment length
0.24 Mean expected error in forward sequences
0.50 Mean expected error in reverse sequences
0.23 Mean expected error in merged sequences
0.18 Mean observed errors in merged region of forward sequences
0.54 Mean observed errors in merged region of reverse sequences
0.72 Mean observed errors in merged region
Merging reads for: COI_100_HK51
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
89943 Pairs
83923 Merged (93.3%)
6020 Not merged (6.7%)
Pairs that failed merging due to various reasons:
41 too few kmers found on same diagonal
2547 too many differences
3423 alignment score too low, or score drop too high
9 staggered read pairs
Statistics of all reads:
250.36 Mean read length
Statistics of merged reads:
364.57 Mean fragment length
9.74 Standard deviation of fragment length
0.26 Mean expected error in forward sequences
0.52 Mean expected error in reverse sequences
0.25 Mean expected error in merged sequences
0.19 Mean observed errors in merged region of forward sequences
0.57 Mean observed errors in merged region of reverse sequences
0.76 Mean observed errors in merged region
Merging reads for: COI_100_HK52
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
80397 Pairs
74255 Merged (92.4%)
6142 Not merged (7.6%)
Pairs that failed merging due to various reasons:
31 too few kmers found on same diagonal
2511 too many differences
3597 alignment score too low, or score drop too high
3 staggered read pairs
Statistics of all reads:
250.38 Mean read length
Statistics of merged reads:
364.59 Mean fragment length
8.22 Standard deviation of fragment length
0.27 Mean expected error in forward sequences
0.44 Mean expected error in reverse sequences
0.23 Mean expected error in merged sequences
0.22 Mean observed errors in merged region of forward sequences
0.46 Mean observed errors in merged region of reverse sequences
0.69 Mean observed errors in merged region
Merging reads for: COI_100_HK53
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
82962 Pairs
76890 Merged (92.7%)
6072 Not merged (7.3%)
Pairs that failed merging due to various reasons:
34 too few kmers found on same diagonal
2217 too many differences
3815 alignment score too low, or score drop too high
6 staggered read pairs
Statistics of all reads:
250.37 Mean read length
Statistics of merged reads:
364.52 Mean fragment length
8.68 Standard deviation of fragment length
0.26 Mean expected error in forward sequences
0.43 Mean expected error in reverse sequences
0.22 Mean expected error in merged sequences
0.19 Mean observed errors in merged region of forward sequences
0.46 Mean observed errors in merged region of reverse sequences
0.65 Mean observed errors in merged region
Merging reads for: COI_100_HK54
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
81013 Pairs
75723 Merged (93.5%)
5290 Not merged (6.5%)
Pairs that failed merging due to various reasons:
38 too few kmers found on same diagonal
2445 too many differences
2802 alignment score too low, or score drop too high
5 staggered read pairs
Statistics of all reads:
250.41 Mean read length
Statistics of merged reads:
364.70 Mean fragment length
7.32 Standard deviation of fragment length
0.24 Mean expected error in forward sequences
0.46 Mean expected error in reverse sequences
0.23 Mean expected error in merged sequences
0.18 Mean observed errors in merged region of forward sequences
0.50 Mean observed errors in merged region of reverse sequences
0.68 Mean observed errors in merged region
Merging reads for: COI_500_HK37
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
120585 Pairs
109424 Merged (90.7%)
11161 Not merged (9.3%)
Pairs that failed merging due to various reasons:
111 too few kmers found on same diagonal
4412 too many differences
6635 alignment score too low, or score drop too high
3 staggered read pairs
Statistics of all reads:
250.44 Mean read length
Statistics of merged reads:
364.34 Mean fragment length
4.63 Standard deviation of fragment length
0.33 Mean expected error in forward sequences
0.47 Mean expected error in reverse sequences
0.25 Mean expected error in merged sequences
0.25 Mean observed errors in merged region of forward sequences
0.49 Mean observed errors in merged region of reverse sequences
0.74 Mean observed errors in merged region
Merging reads for: COI_500_HK38
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
93778 Pairs
86827 Merged (92.6%)
6951 Not merged (7.4%)
Pairs that failed merging due to various reasons:
88 too few kmers found on same diagonal
2664 too many differences
4191 alignment score too low, or score drop too high
8 staggered read pairs
Statistics of all reads:
250.52 Mean read length
Statistics of merged reads:
364.40 Mean fragment length
4.26 Standard deviation of fragment length
0.30 Mean expected error in forward sequences
0.48 Mean expected error in reverse sequences
0.24 Mean expected error in merged sequences
0.21 Mean observed errors in merged region of forward sequences
0.49 Mean observed errors in merged region of reverse sequences
0.71 Mean observed errors in merged region
Merging reads for: COI_500_HK39
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
146550 Pairs
134723 Merged (91.9%)
11827 Not merged (8.1%)
Pairs that failed merging due to various reasons:
156 too few kmers found on same diagonal
1 multiple potential alignments
4664 too many differences
6996 alignment score too low, or score drop too high
10 staggered read pairs
Statistics of all reads:
250.45 Mean read length
Statistics of merged reads:
364.09 Mean fragment length
8.67 Standard deviation of fragment length
0.32 Mean expected error in forward sequences
0.45 Mean expected error in reverse sequences
0.24 Mean expected error in merged sequences
0.24 Mean observed errors in merged region of forward sequences
0.47 Mean observed errors in merged region of reverse sequences
0.71 Mean observed errors in merged region
Merging reads for: COI_500_HK40
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
119622 Pairs
109897 Merged (91.9%)
9725 Not merged (8.1%)
Pairs that failed merging due to various reasons:
128 too few kmers found on same diagonal
3864 too many differences
5728 alignment score too low, or score drop too high
5 staggered read pairs
Statistics of all reads:
250.48 Mean read length
Statistics of merged reads:
364.28 Mean fragment length
5.43 Standard deviation of fragment length
0.31 Mean expected error in forward sequences
0.49 Mean expected error in reverse sequences
0.24 Mean expected error in merged sequences
0.23 Mean observed errors in merged region of forward sequences
0.50 Mean observed errors in merged region of reverse sequences
0.73 Mean observed errors in merged region
Merging reads for: COI_500_HK41
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
128094 Pairs
117560 Merged (91.8%)
10534 Not merged (8.2%)
Pairs that failed merging due to various reasons:
179 too few kmers found on same diagonal
4244 too many differences
6104 alignment score too low, or score drop too high
7 staggered read pairs
Statistics of all reads:
250.51 Mean read length
Statistics of merged reads:
364.37 Mean fragment length
6.25 Standard deviation of fragment length
0.27 Mean expected error in forward sequences
0.48 Mean expected error in reverse sequences
0.24 Mean expected error in merged sequences
0.19 Mean observed errors in merged region of forward sequences
0.54 Mean observed errors in merged region of reverse sequences
0.72 Mean observed errors in merged region
Merging reads for: COI_500_HK42
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
84467 Pairs
77020 Merged (91.2%)
7447 Not merged (8.8%)
Pairs that failed merging due to various reasons:
69 too few kmers found on same diagonal
1 multiple potential alignments
3141 too many differences
4233 alignment score too low, or score drop too high
3 staggered read pairs
Statistics of all reads:
250.61 Mean read length
Statistics of merged reads:
364.63 Mean fragment length
6.61 Standard deviation of fragment length
0.24 Mean expected error in forward sequences
0.53 Mean expected error in reverse sequences
0.24 Mean expected error in merged sequences
0.17 Mean observed errors in merged region of forward sequences
0.64 Mean observed errors in merged region of reverse sequences
0.80 Mean observed errors in merged region
Merging reads for: COI_500_HK49
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
74265 Pairs
67718 Merged (91.2%)
6547 Not merged (8.8%)
Pairs that failed merging due to various reasons:
81 too few kmers found on same diagonal
2989 too many differences
3473 alignment score too low, or score drop too high
4 staggered read pairs
Statistics of all reads:
250.47 Mean read length
Statistics of merged reads:
364.75 Mean fragment length
6.74 Standard deviation of fragment length
0.24 Mean expected error in forward sequences
0.56 Mean expected error in reverse sequences
0.26 Mean expected error in merged sequences
0.17 Mean observed errors in merged region of forward sequences
0.62 Mean observed errors in merged region of reverse sequences
0.79 Mean observed errors in merged region
Merging reads for: COI_500_HK50
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
81354 Pairs
75588 Merged (92.9%)
5766 Not merged (7.1%)
Pairs that failed merging due to various reasons:
81 too few kmers found on same diagonal
2482 too many differences
3199 alignment score too low, or score drop too high
4 staggered read pairs
Statistics of all reads:
250.52 Mean read length
Statistics of merged reads:
364.66 Mean fragment length
7.57 Standard deviation of fragment length
0.27 Mean expected error in forward sequences
0.47 Mean expected error in reverse sequences
0.23 Mean expected error in merged sequences
0.20 Mean observed errors in merged region of forward sequences
0.49 Mean observed errors in merged region of reverse sequences
0.70 Mean observed errors in merged region
Merging reads for: COI_500_HK51
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
113932 Pairs
105503 Merged (92.6%)
8429 Not merged (7.4%)
Pairs that failed merging due to various reasons:
110 too few kmers found on same diagonal
3809 too many differences
4505 alignment score too low, or score drop too high
5 staggered read pairs
Statistics of all reads:
250.54 Mean read length
Statistics of merged reads:
364.66 Mean fragment length
7.14 Standard deviation of fragment length
0.26 Mean expected error in forward sequences
0.52 Mean expected error in reverse sequences
0.24 Mean expected error in merged sequences
0.20 Mean observed errors in merged region of forward sequences
0.54 Mean observed errors in merged region of reverse sequences
0.74 Mean observed errors in merged region
Merging reads for: COI_500_HK52
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
121246 Pairs
113930 Merged (94.0%)
7316 Not merged (6.0%)
Pairs that failed merging due to various reasons:
91 too few kmers found on same diagonal
2602 too many differences
4619 alignment score too low, or score drop too high
4 staggered read pairs
Statistics of all reads:
250.43 Mean read length
Statistics of merged reads:
364.74 Mean fragment length
6.49 Standard deviation of fragment length
0.21 Mean expected error in forward sequences
0.44 Mean expected error in reverse sequences
0.20 Mean expected error in merged sequences
0.15 Mean observed errors in merged region of forward sequences
0.46 Mean observed errors in merged region of reverse sequences
0.61 Mean observed errors in merged region
Merging reads for: COI_500_HK53
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
108046 Pairs
100604 Merged (93.1%)
7442 Not merged (6.9%)
Pairs that failed merging due to various reasons:
104 too few kmers found on same diagonal
2833 too many differences
4502 alignment score too low, or score drop too high
3 staggered read pairs
Statistics of all reads:
250.43 Mean read length
Statistics of merged reads:
364.62 Mean fragment length
6.46 Standard deviation of fragment length
0.23 Mean expected error in forward sequences
0.43 Mean expected error in reverse sequences
0.22 Mean expected error in merged sequences
0.18 Mean observed errors in merged region of forward sequences
0.47 Mean observed errors in merged region of reverse sequences
0.64 Mean observed errors in merged region
Merging reads for: COI_500_HK54
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
104984 Pairs
95343 Merged (90.8%)
9641 Not merged (9.2%)
Pairs that failed merging due to various reasons:
74 too few kmers found on same diagonal
2427 too many differences
7134 alignment score too low, or score drop too high
6 staggered read pairs
Statistics of all reads:
250.33 Mean read length
Statistics of merged reads:
364.39 Mean fragment length
10.29 Standard deviation of fragment length
0.23 Mean expected error in forward sequences
0.44 Mean expected error in reverse sequences
0.21 Mean expected error in merged sequences
0.17 Mean observed errors in merged region of forward sequences
0.46 Mean observed errors in merged region of reverse sequences
0.63 Mean observed errors in merged region
Merging reads for: NTC_1
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
960 Pairs
306 Merged (31.9%)
654 Not merged (68.1%)
Pairs that failed merging due to various reasons:
13 too few kmers found on same diagonal
134 too many differences
501 alignment score too low, or score drop too high
6 staggered read pairs
Statistics of all reads:
233.35 Mean read length
Statistics of merged reads:
287.81 Mean fragment length
132.89 Standard deviation of fragment length
0.49 Mean expected error in forward sequences
1.03 Mean expected error in reverse sequences
0.63 Mean expected error in merged sequences
0.56 Mean observed errors in merged region of forward sequences
1.49 Mean observed errors in merged region of reverse sequences
2.05 Mean observed errors in merged region
Merging reads for: NTC_2
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
10408 Pairs
7842 Merged (75.3%)
2566 Not merged (24.7%)
Pairs that failed merging due to various reasons:
1590 too few kmers found on same diagonal
15 multiple potential alignments
109 too many differences
574 alignment score too low, or score drop too high
278 staggered read pairs
Statistics of all reads:
80.31 Mean read length
Statistics of merged reads:
64.72 Mean fragment length
42.73 Standard deviation of fragment length
0.09 Mean expected error in forward sequences
0.11 Mean expected error in reverse sequences
0.05 Mean expected error in merged sequences
0.18 Mean observed errors in merged region of forward sequences
0.25 Mean observed errors in merged region of reverse sequences
0.43 Mean observed errors in merged region
Merging reads for: NTC_3
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Merging reads 100%
7779 Pairs
6114 Merged (78.6%)
1665 Not merged (21.4%)
Pairs that failed merging due to various reasons:
413 too few kmers found on same diagonal
10 multiple potential alignments
162 too many differences
819 alignment score too low, or score drop too high
261 staggered read pairs
Statistics of all reads:
93.17 Mean read length
Statistics of merged reads:
72.01 Mean fragment length
52.72 Standard deviation of fragment length
0.12 Mean expected error in forward sequences
0.14 Mean expected error in reverse sequences
0.09 Mean expected error in merged sequences
0.28 Mean observed errors in merged region of forward sequences
0.30 Mean observed errors in merged region of reverse sequences
0.58 Mean observed errors in merged region
When executing this for loop, VSEARCH will attempt to merge all reads from the forward and reverse sequencing file for each sample separately. The output files containing the merged reads will be placed in the subdirectory sequenceData/2-raw/ and will have the extension _merged.fastq rather than _1.fastq or _2.fastq. Within the Terminal window, all the statistics are reported for each sample separately. A quick glance shows us that we managed to merge roughly 90% of reads for each sample. However, due to the number of samples, it is difficult to get an overview of the read merging success rate and compare different samples.
2.3 Summarizing the output#
To get a better overview, we can use the following python script to generate a bar plot with the raw and merged read statistics. The python code takes in two arguments, the first is the location where the merged sequence files are stored, the second is the location of the raw sequence files. Since this is python code, we cannot directly copy-paste the code into the Terminal as we did before. Rather, we have to create our first script. To do this, we will use a text editor that is implemented in the Terminal called nano. Running nano will open a new text window where we can copy-paste our python code. For this tutorial, it is not important or essential to understand python, though it is an extremely powerful and useful coding language for bioinforamticians!. To provide some context, I’ll give a brief explanation during the workshop when we’re executing this code.
nano sequenceData/1-scripts/rawMergedStatistics.py
#! /usr/bin/env python3
## import modules
import os
import sys
import matplotlib.pyplot as plt
import pandas as pd
import seaborn as sns
## user arguments
mergedPath = sys.argv[1]
rawPath = sys.argv[2]
## first, create a sample name list
mergedFileList = os.listdir(mergedPath)
sampleNameList = []
for mergedFile in mergedFileList:
sampleName = mergedFile.split('_merged.fastq')[0]
if sampleName.startswith('COI_') or sampleName.startswith('NTC_'):
sampleNameList.append(sampleName)
## count number of raw and merged sequences for each sample in sampleNameList
rawSeqCount = {}
mergedSeqCount = {}
for sample in sampleNameList:
with open(f'{mergedPath}{sample}_merged.fastq', 'r') as mergedFile:
x = len(mergedFile.readlines()) / 4
mergedSeqCount[sample] = int(x)
with open(f'{rawPath}{sample}_1.fastq', 'r') as rawFile:
y = len(rawFile.readlines()) / 4
rawSeqCount[sample] = int(y)
## create a dataframe from the dictionaries
df = pd.DataFrame({'Sample': list(rawSeqCount.keys()), 'Raw': list(rawSeqCount.values()), 'Merged': list(mergedSeqCount.values())})
## sort the dataframe by raw reads in descending order
df = df.sort_values(by='Raw', ascending=False)
## calculate the percentage of merged/raw and format it with 2 decimal places and the '%' symbol
df['Percentage'] = (df['Merged'] / df['Raw'] * 100).round(2).astype(str) + '%'
## create a horizontal bar plot using seaborn
plt.figure(figsize=(20, 8)) # Adjust the figure size as needed
## use seaborn's barplot
ax = sns.barplot(x='Raw', y='Sample', data=df, label='Raw', color='#BBC6C8')
sns.barplot(x='Merged', y='Sample', data=df, label='Merged', color='#469597')
## add labels and title
plt.xlabel('Number of sequences')
plt.ylabel('Samples')
plt.title('Horizontal bar graph of raw and merged reads (Sorted by Total in Reverse)')
## add a legend
plt.legend()
# Add raw read count next to the bars
for i, v in enumerate(df['Percentage']):
ax.text(df['Raw'].values[i] + 50, i, v, va='center', fontsize=10, color='black')
## save the plot
plt.tight_layout()
plt.savefig('raw_and_merged_bargraph.png', dpi = 300)
Once we have copy-pasted the code, we can press ctrl +x to exit out of the editor, followed by y and return to save the file. After doing so, we’re back in the normal Terminal window. Before we can run or execute our first script, we need to make it executable.
Important
Remember the permissions for each file that we discussed before? By running the ls -ltr command, we can see that for the script we have just created, we only have read (r) and write (w) access, but no execution (x) permission. This is essential in this case, as we won’t be running the script using the sbatch command.
ls -ltr sequenceData/1-scripts/
Output
-rw-r--r-- 1 ednaw01 others 776 Oct 21 10:59 example-script.sh
-rw-r--r-- 1 ednaw01 others 169 Oct 21 10:59 module_load
-rw-r--r-- 1 ednaw01 others 833 Oct 21 11:33 fastqc_raw.sh
-rw-r--r-- 1 ednaw01 others 822 Oct 21 12:03 multiqc_raw.sh
-rw-r--r-- 1 ednaw01 others 955 Oct 21 12:10 merge_single.sh
-rw-r--r-- 1 ednaw01 others 985 Oct 21 12:21 merge_batch.sh
-rw-r--r-- 1 ednaw01 others 2146 Oct 21 12:26 rawMergedStatistics.py
To change the permissions or modifiers of a file, we can use the chmod command, which stands for change modifier. Since we want to make our file executable, we can specify the parameter +x.
chmod +x sequenceData/1-scripts/rawMergedStatistics.py
Rerunning the ls -ltr command shows that we have changed the permissions and that we can execute our script.
ls -ltr sequenceData/1-scripts/
Output
-rw-r--r-- 1 ednaw01 others 776 Oct 21 10:59 example-script.sh
-rw-r--r-- 1 ednaw01 others 169 Oct 21 10:59 module_load
-rw-r--r-- 1 ednaw01 others 833 Oct 21 11:33 fastqc_raw.sh
-rw-r--r-- 1 ednaw01 others 822 Oct 21 12:03 multiqc_raw.sh
-rw-r--r-- 1 ednaw01 others 955 Oct 21 12:10 merge_single.sh
-rw-r--r-- 1 ednaw01 others 985 Oct 21 12:21 merge_batch.sh
-rwxr-xr-x 1 ednaw01 others 2146 Oct 21 12:26 rawMergedStatistics.py
To execute the script, we can use the ./ command followed by the python script and our two user parameters where our (parameter 1) merged and (parameter 2) raw files are located.
nano sequenceData/1-scripts/example-script.sh
./sequenceData/1-scripts/rawMergedStatistics.py sequenceData/2-raw/ sequenceData/2-raw/unzipped/
Press ctrl + x and y to save the file and change the name to check_merging.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/check_merging.sh
scp -r ednaw01@hpc2021-io1.hku.hk:~/ednaw01/raw_and_merged_bargraph.png ./
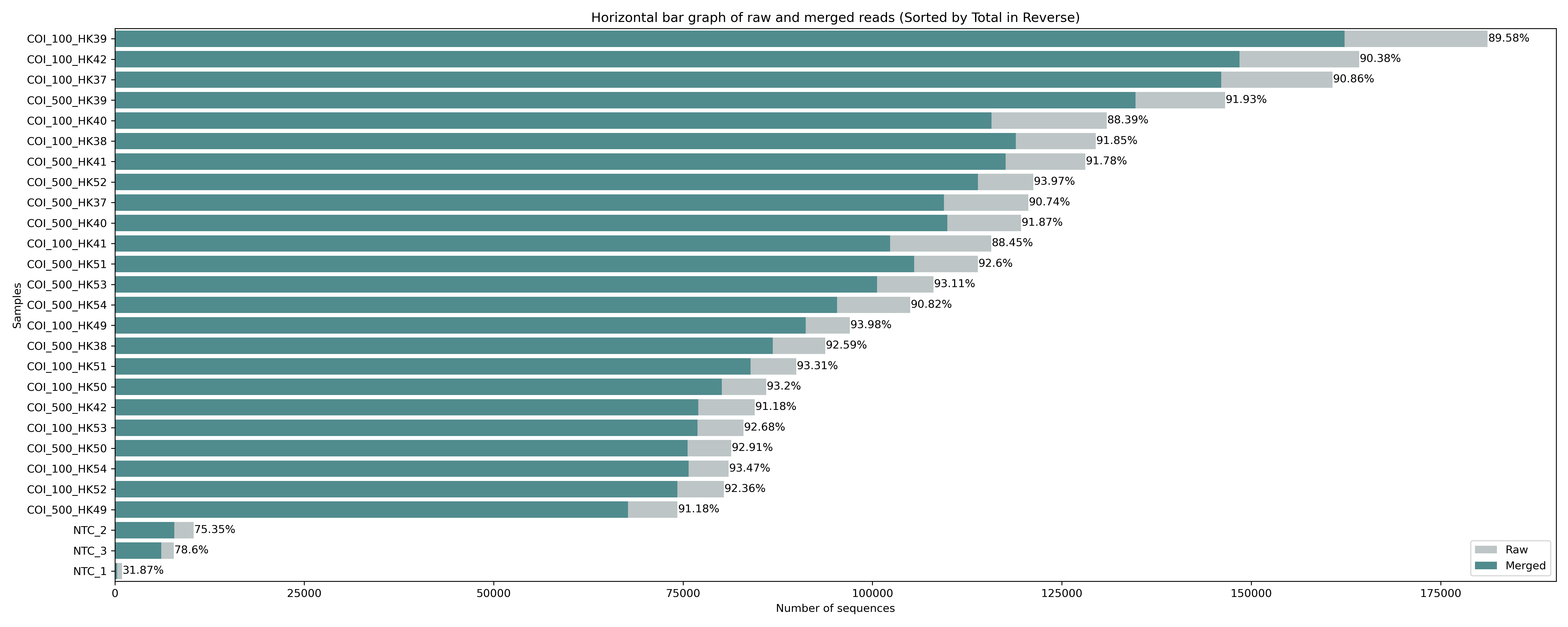
Fig. 10 : Raw and merged read count#
The figure we generated using the python script shows a horizontal bar graph with the merged reads in green and the raw sequence counts in grey. We can see that the percentage of merged reads is similar across all the samples, roughly 90%. We can also see that there is some abundance differences between the samples, which is to be expected, as pooling samples equimolarly is hard to do in the lab, something we covered during the first week of this eDNA workshop. Also, note the big difference between the samples and the negative controls, something we observed earlier in the MULTIQC report as well.
Amplicon length exceeding cycle number
To merge forward and reverse reads, it is essential to have at least a partial overlap in the amplicon region with the forward and reverse reads. Even better would be a full overlap, as it increases the base call quality score. Sometimes, however, the amplicon length is too long to achieve partial overlap with paired-end reads on an Illumina platform, since the Illumina sequencing technology is restricted by sequence length. While such a scenario should be avoided at all cost, if this was a situation you would find yourself in, you can concatenate paired-end reads using the following line of code, rather than merge them.
vsearch --fastq_join input_R1.fastq --reverse input_R2.fastq --fastqout output_joined.fastq
Keep in mind 3rd generation sequencing technologies, which we discussed during the first week of this workshop, if your experiment would benefit from using longer amplicon lengths than can be covered by Illumina sequencing technology.
3. Removing primer sequences#
3.1 A single sample example#
At this point, our merged reads still contain the primer sequences. Since these regions are artefacts from the PCR amplification and not biological, they will need to be removed from the reads before continuing with the bioinformatic pipeline. For this library and experiment, we have used the mlCOIintF/jgHCO2198 primer set (Leray et al., 2013). The forward primer corresponds to 5’-GGWACWGGWTGAACWGTWTAYCCYCC-3’ and the reverse primer sequence is 5’-TAIACYTCIGGRTGICCRAARAAYCA-3’. Before batch processing every sample, let’s test our code on a single sample again to start with. For primer or adapter removal, we can use the program cutadapt. To specify the primers to be removed, we can use the -a parameter. Since we’re removing both the forward and reverse primer, we can link them together using .... Remember to use the reverse complement of the reverse primer, as this would be the direction the reverse primer is found in our sequence data after merging. The minimum and maximum length of the amplicon can be specified with the -m and -M parameters, respectively. To only keep reads for which both primers were found and removed, we need to specify the --discard-untrimmed option. The --no-indels and -e 2 parameters allow us to tell the program to not include insertions and deletions in the search and allow a maximum of 2 errors in the primer sequence. We can specify the --revcomp parameter to search for the primer sequences in both directions. Finally, we can use the --cores=0 parameter to automatically detect the number of available cores.
nano sequenceData/1-scripts/example-script.sh
cutadapt sequenceData/2-raw/COI_100_HK37_merged.fastq -a GGWACWGGWTGAACWGTWTAYCCYCC...TGRTTYTTYGGHCAYCCHGARGTHTA -m 300 -M 330 --discard-untrimmed -o sequenceData/4-demux/COI_100_HK37_trimmed.fastq --no-indels -e 2 --revcomp --cores=0
Press ctrl + x and y to save the file and change the name to adapter_removal_single.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/adapter_removal_single.sh
Output
This is cutadapt 4.4 with Python 3.11.5
Command line parameters: sequenceData/2-raw/COI_100_HK37_merged.fastq -a GGWACWGGWTGAACWGTWTAYCCYCC...TGRTTYTTYGGHCAYCCHGARGTHTA -m 300 -M 330 --discard-untrimmed -o sequenceData/4-demux/COI_100_HK37_trimmed.fastq --no-indels -e 2 --revcomp --cores=0
Processing single-end reads on 8 cores ...
Done 00:00:00 146,040 reads @ 4.1 µs/read; 14.69 M reads/minute
Finished in 0.598 s (4.093 µs/read; 14.66 M reads/minute).
=== Summary ===
Total reads processed: 146,040
Reads with adapters: 145,999 (100.0%)
Reverse-complemented: 4 (0.0%)
== Read fate breakdown ==
Reads that were too short: 4,877 (3.3%)
Reads that were too long: 4,754 (3.3%)
Reads discarded as untrimmed: 0 (0.0%)
Reads written (passing filters): 136,409 (93.4%)
Total basepairs processed: 52,491,397 bp
Total written (filtered): 42,623,291 bp (81.2%)
=== Adapter 1 ===
Sequence: GGWACWGGWTGAACWGTWTAYCCYCC...TGRTTYTTYGGHCAYCCHGARGTHTA; Type: linked; Length: 26+26; 5' trimmed: 143311 times; 3' trimmed: 143808 times; Reverse-complemented: 4 times
Minimum overlap: 3+3
No. of allowed errors:
1-12 bp: 0; 13-25 bp: 1; 26 bp: 2
No. of allowed errors:
1-12 bp: 0; 13-25 bp: 1; 26 bp: 2
Overview of removed sequences at 5' end
length count expect max.err error counts
4 2 570.5 0 2
5 1 142.6 0 1
11 1 0.0 0 1
13 1 0.0 1 1
14 1 0.0 1 0 1
15 6 0.0 1 6
16 5 0.0 1 4 1
17 10 0.0 1 2 8
18 5 0.0 1 4 1
19 20 0.0 1 14 6
20 13 0.0 1 11 2
21 15 0.0 1 6 9
22 12 0.0 1 10 2
23 10 0.0 1 9 1
24 31 0.0 1 14 17
25 851 0.0 1 552 299
26 142152 0.0 2 140479 1461 212
27 175 0.0 2 142 27 6
Overview of removed sequences at 3' end
length count expect max.err error counts
13 1 0.0 1 0 1
14 2 0.0 1 2
15 1 0.0 1 1
16 5 0.0 1 2 3
17 3 0.0 1 0 3
18 10 0.0 1 4 6
19 3 0.0 1 0 3
20 6 0.0 1 4 2
21 14 0.0 1 9 5
22 19 0.0 1 15 4
23 65 0.0 1 48 17
24 77 0.0 1 62 15
25 174 0.0 1 45 129
26 142067 0.0 2 140032 1686 349
27 1351 0.0 2 749 370 232
28 8 0.0 2 3 2 3
63 1 0.0 2 0 0 1
70 1 0.0 2 0 0 1
The cutadapt output provides information on how many reads were analysed, how many reads were trimmed on the plus and minus strand, plus a detailed overview of where the adapters were cut in the sequence. For our sample sequenceData/2-raw/COI_100_HK37_merged.fastq, 146,040 reads were processed, 145,999 reads were found to contain both primers, and 4 reads were found where the primers were present as the reverse complement of the read. As these options that we specified in our command managed to remove the primer sequences from nearly all reads, we will use these settings in our for loop to batch process all samples.
3.2 batch trimming#
Similarly to the merging of reads, we need to process 27 samples. Hence, we will write another for loop to accomplish this goal, rather than executing the code to trim primer sequences 27 times by ourselves. Because the cutadapt output that is written to the Terminal window is very elaborate, we will write it to a file instead. In the next section (Summarizing cutadapt output), we’ll parse the output to produce some graphs in order to get a better overview of how cutadapt performed across the 27 samples. First, we will remove the file we just generated and move to the sequenceData/2-raw/ directory. As we’ve introduced the for loop syntax before and explained the cutadapt code snippet, I won’t be going into too much detail on each aspect.
rm sequenceData/4-demux/COI_100_HK37_trimmed.fastq
nano sequenceData/1-scripts/example-script.sh
cd sequenceData/2-raw/
for fq in *merged.fastq
do
echo "trimming primer seqs for: ${fq/_merged.fastq/}"
cutadapt ${fq} -a GGWACWGGWTGAACWGTWTAYCCYCC...TGRTTYTTYGGHCAYCCHGARGTHTA -m 300 -M 330 --discard-untrimmed -o ../4-demux/${fq/merged.fastq/trimmed.fastq} --no-indels -e 2 --revcomp --cores=0 >> ../0-metadata/cutadapt_primer_trimming.txt
done
Press ctrl + x and y to save the file and change the name to adapter_removal_batch.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/adapter_removal_batch.sh
Output
trimming primer seqs for: COI_100_HK37
Done 00:00:00 146,040 reads @ 3.4 µs/read; 17.58 M reads/minute
trimming primer seqs for: COI_100_HK38
Done 00:00:00 118,917 reads @ 3.7 µs/read; 16.14 M reads/minute
trimming primer seqs for: COI_100_HK39
Done 00:00:00 162,326 reads @ 3.5 µs/read; 17.39 M reads/minute
trimming primer seqs for: COI_100_HK40
Done 00:00:00 115,722 reads @ 3.8 µs/read; 15.94 M reads/minute
trimming primer seqs for: COI_100_HK41
Done 00:00:00 102,320 reads @ 3.8 µs/read; 15.74 M reads/minute
trimming primer seqs for: COI_100_HK42
Done 00:00:00 148,463 reads @ 3.6 µs/read; 16.78 M reads/minute
trimming primer seqs for: COI_100_HK49
Done 00:00:00 91,171 reads @ 4.1 µs/read; 14.62 M reads/minute
trimming primer seqs for: COI_100_HK50
Done 00:00:00 80,114 reads @ 4.3 µs/read; 14.11 M reads/minute
trimming primer seqs for: COI_100_HK51
Done 00:00:00 83,923 reads @ 4.7 µs/read; 12.89 M reads/minute
trimming primer seqs for: COI_100_HK52
Done 00:00:00 74,255 reads @ 4.2 µs/read; 14.24 M reads/minute
trimming primer seqs for: COI_100_HK53
Done 00:00:00 76,890 reads @ 4.3 µs/read; 14.04 M reads/minute
trimming primer seqs for: COI_100_HK54
Done 00:00:00 75,723 reads @ 4.3 µs/read; 14.11 M reads/minute
trimming primer seqs for: COI_500_HK37
Done 00:00:00 109,424 reads @ 3.7 µs/read; 16.40 M reads/minute
trimming primer seqs for: COI_500_HK38
Done 00:00:00 86,827 reads @ 4.3 µs/read; 13.98 M reads/minute
trimming primer seqs for: COI_500_HK39
Done 00:00:00 134,723 reads @ 3.9 µs/read; 15.53 M reads/minute
trimming primer seqs for: COI_500_HK40
Done 00:00:00 109,897 reads @ 4.0 µs/read; 15.04 M reads/minute
trimming primer seqs for: COI_500_HK41
Done 00:00:00 117,560 reads @ 4.0 µs/read; 14.88 M reads/minute
trimming primer seqs for: COI_500_HK42
Done 00:00:00 77,020 reads @ 5.1 µs/read; 11.87 M reads/minute
trimming primer seqs for: COI_500_HK49
Done 00:00:00 67,718 reads @ 6.3 µs/read; 9.52 M reads/minute
trimming primer seqs for: COI_500_HK50
Done 00:00:00 75,588 reads @ 5.2 µs/read; 11.48 M reads/minute
trimming primer seqs for: COI_500_HK51
Done 00:00:00 105,503 reads @ 4.4 µs/read; 13.75 M reads/minute
trimming primer seqs for: COI_500_HK52
Done 00:00:00 113,930 reads @ 4.0 µs/read; 15.13 M reads/minute
trimming primer seqs for: COI_500_HK53
Done 00:00:00 100,604 reads @ 4.3 µs/read; 14.01 M reads/minute
trimming primer seqs for: COI_500_HK54
Done 00:00:00 95,343 reads @ 4.3 µs/read; 13.86 M reads/minute
trimming primer seqs for: NTC_1
Done 00:00:00 306 reads @ 435.8 µs/read; 0.14 M reads/minute
trimming primer seqs for: NTC_2
Done 00:00:00 7,842 reads @ 21.1 µs/read; 2.85 M reads/minute
trimming primer seqs for: NTC_3
Done 00:00:00 6,114 reads @ 24.3 µs/read; 2.47 M reads/minute
When executing this for loop, cutadapt will attempt to remove the primer sequences from all reads in both directions (parameter --revcomp). The output files containing the reads where the primers were successfully removed are placed in the subdirectory sequenceData/4-demux/ and will have the extension _trimmed.fastq rather than _merged.fastq. As mentioned above, the cutadapt output was written to sequenceData/0-metadata/cutadapt_primer_trimming.txt, rather than the Terminal window. We can open this text file using a text editor, such as Sublime Text. However, due to the number of samples, it is difficult to get an overview of the success rate and compare the different samples, a similar issue we encountered when merging forward and reverse reads.
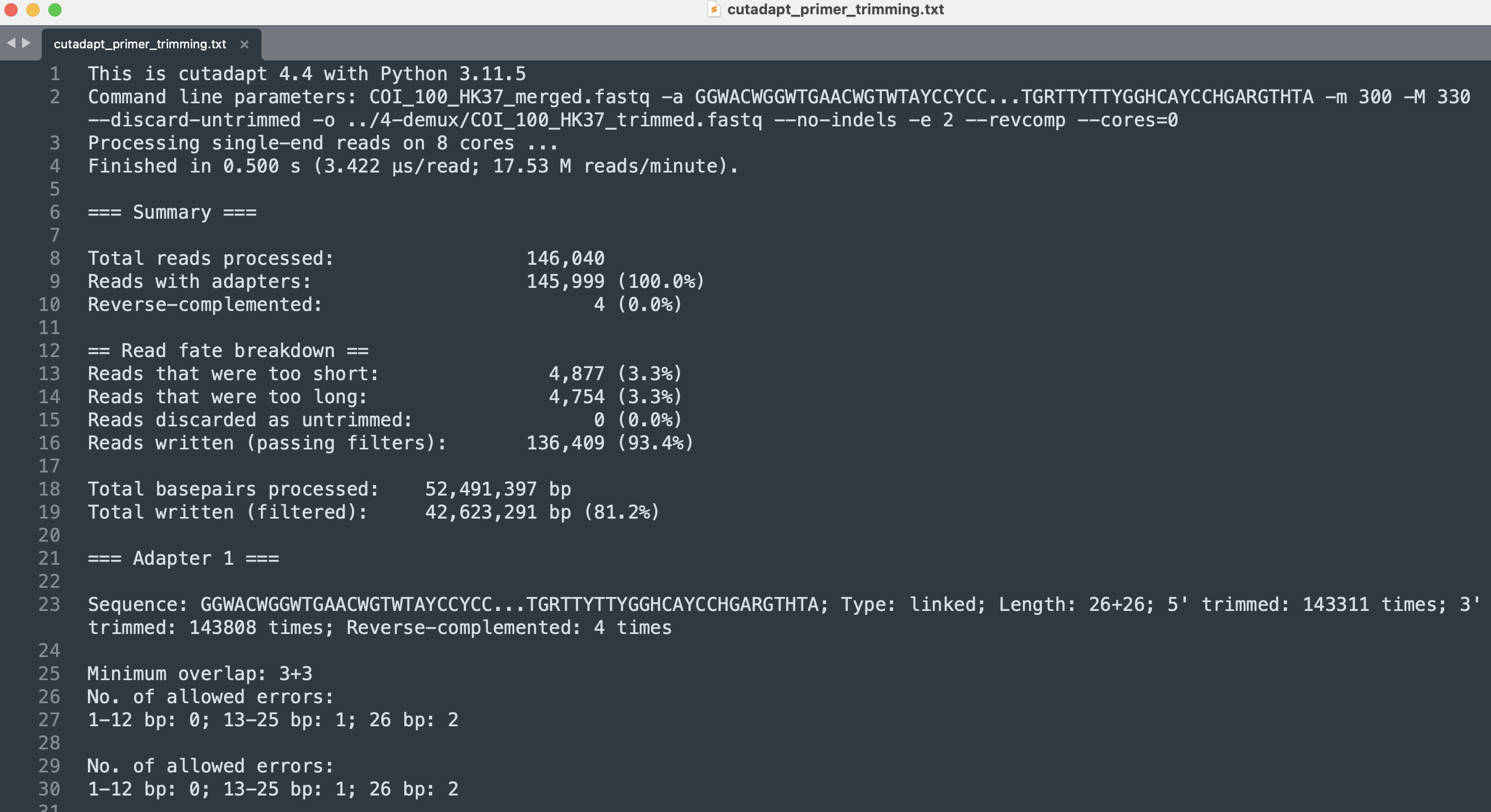
Fig. 11 : Output of the cutadapt code, providing information about adapter removal success rate.#
3.3 Summarizing cutadapt output#
To get a better overview of the read statistics after primer trimming and the success rate of cutadapt to locate and trim the primer-binding regions, we can alter the python code producing the bar graph to incorporate the sequence counts after primer trimming. This new version of the code takes in three arguments, the first is the location where the merged sequence files are stored, the second is the location of the raw sequence files, and the third is the location of the trimmed sequence files. Rather than trying to alter the code in the already-existing script sequenceData/1-scripts/rawMergedStatistics.py, we will generate a new script where we can simply cut and paste the code below. Generating, saving, and making the script executable follows the same process as discussed previously.
nano sequenceData/1-scripts/rawMergedTrimmedStatistics.py
#! /usr/bin/env python3
## import modules
import os
import sys
import matplotlib.pyplot as plt
import pandas as pd
import seaborn as sns
## user arguments
mergedPath = sys.argv[1]
rawPath = sys.argv[2]
trimmedPath = sys.argv[3]
## first, create a sample name list
mergedFileList = os.listdir(mergedPath)
sampleNameList = []
for mergedFile in mergedFileList:
sampleName = mergedFile.split('_merged.fastq')[0]
if sampleName.startswith('COI_') or sampleName.startswith('NTC_'):
sampleNameList.append(sampleName)
## count number of raw and merged sequences for each sample in sampleNameList
rawSeqCount = {}
mergedSeqCount = {}
trimmedSeqCount = {}
for sample in sampleNameList:
with open(f'{mergedPath}{sample}_merged.fastq', 'r') as mergedFile:
x = len(mergedFile.readlines()) / 4
mergedSeqCount[sample] = int(x)
with open(f'{rawPath}{sample}_1.fastq', 'r') as rawFile:
y = len(rawFile.readlines()) / 4
rawSeqCount[sample] = int(y)
with open(f'{trimmedPath}{sample}_trimmed.fastq', 'r') as trimmedFile:
z = len(trimmedFile.readlines()) / 4
trimmedSeqCount[sample] = int(z)
## create a dataframe from the dictionaries
df = pd.DataFrame({'Sample': list(rawSeqCount.keys()), 'Raw': list(rawSeqCount.values()), 'Merged': list(mergedSeqCount.values()), 'Trimmed': list(trimmedSeqCount.values())})
## sort the dataframe by raw reads in descending order
df = df.sort_values(by='Raw', ascending=False)
## calculate the percentage of merged/raw and format it with 2 decimal places and the '%' symbol
df['Percentage'] = (df['Trimmed'] / df['Raw'] * 100).round(2).astype(str) + '%'
## create a horizontal bar plot using seaborn
plt.figure(figsize=(20, 8)) # Adjust the figure size as needed
## use seaborn's barplot
ax = sns.barplot(x='Raw', y='Sample', data=df, label='Raw', color='#BBC6C8')
sns.barplot(x='Merged', y='Sample', data=df, label='Merged', color='#469597')
sns.barplot(x='Trimmed', y='Sample', data=df, label='Trimmed', color='#DDBEAA')
## add labels and title
plt.xlabel('Number of sequences')
plt.ylabel('Samples')
plt.title('Horizontal bar graph of raw, merged, and trimmed reads (Sorted by Total in Reverse)')
## add a legend
plt.legend()
# Add raw read count next to the bars
for i, v in enumerate(df['Percentage']):
ax.text(df['Raw'].values[i] + 50, i, v, va='center', fontsize=10, color='black')
## save the plot
plt.tight_layout()
plt.savefig('raw_and_merged_and_trimmed_bargraph.png', dpi = 300)
Press ctrl +x to exit out of the editor, followed by y and return.
chmod +x sequenceData/1-scripts/rawMergedTrimmedStatistics.py
nano sequenceData/1-scripts/example-script.sh
./sequenceData/1-scripts/rawMergedTrimmedStatistics.py sequenceData/2-raw/ sequenceData/2-raw/unzipped/ sequenceData/4-demux/
Press ctrl + x and y to save the file and change the name to check_adapter_trimming.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/check_adapter_trimming.sh
scp -r ednaw01@hpc2021-io1.hku.hk:~/ednaw01/raw_and_merged_and_trimmed_bargraph.png ./
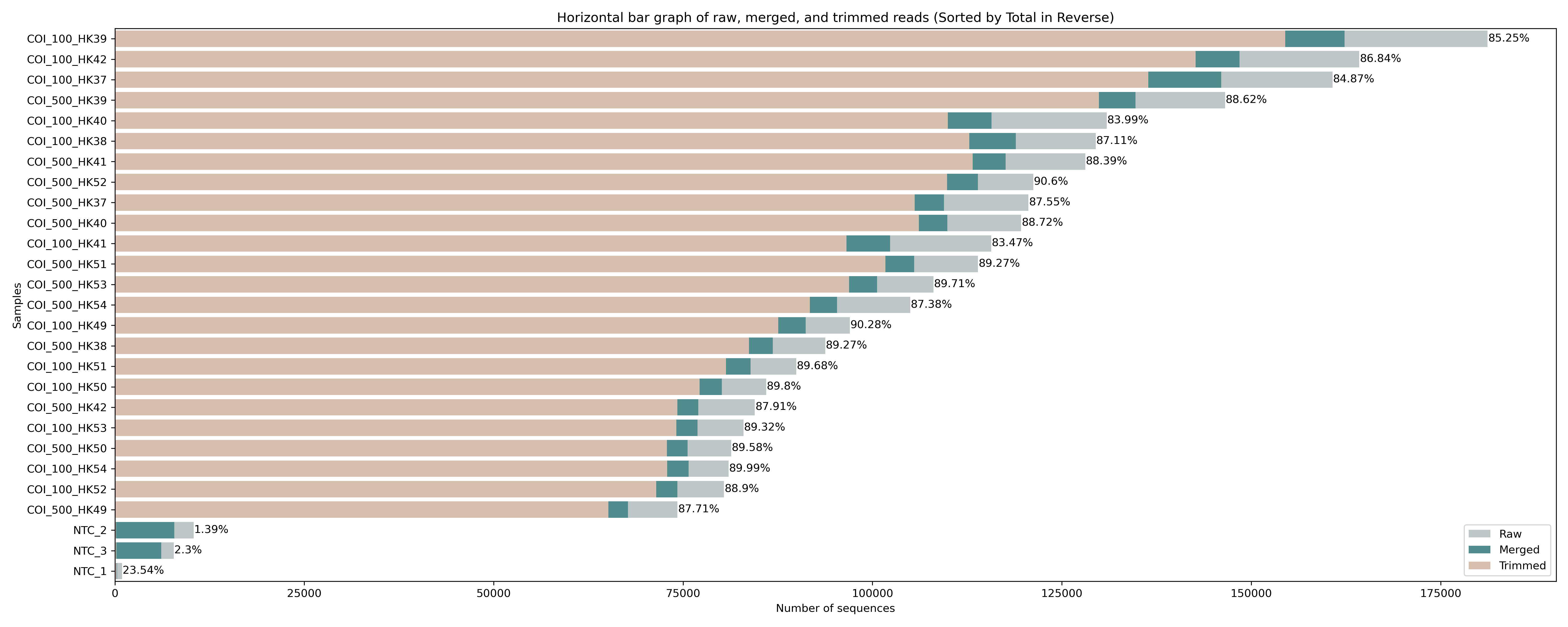
Fig. 12 : Read count of raw, merged, and trimmed files.#
The figure shows a similar success rate for cutadapt to locate and trim primers across all samples, with roughly 85% of raw sequences still included after merging and primer trimming. Interestingly, for the negative control samples (NTC_1, NTC_2, and NTC_3) hardly any reads are returned after primer trimming. To see what happened with the negative controls, let’s run this little python code below to print out the cutadapt summary from the file we generated during the for loop. The python code takes two user arguments, which are (1) the file name of the cutadapt summary file to parse and (2) a list of sample names to print the summary statistics for. This list should be separated by + symbols.
nano sequenceData/1-scripts/cutadaptParsing.py
#! /usr/bin/env python3
## import modules
import sys
## user arguments
cutadaptResults = sys.argv[1]
sampleNames = sys.argv[2]
## create sample list
sampleList = sampleNames.split('+')
## parse cutadapt file
printLine = 0
startPrint = 0
with open(cutadaptResults, 'r') as infile:
for line in infile:
if line.startswith('=== Adapter 1 ==='):
startPrint = 0
printLine = 0
if startPrint == 1:
print(line.rstrip('\n'))
if printLine == 1:
if line.startswith('=== Summary ==='):
startPrint = 1
print(line.rstrip('\n'))
if line.startswith('Command line parameters:'):
for sample in sampleList:
if sample in line:
print(f'{sample} summary statistics:')
printLine = 1
Press ctrl +x to exit out of the editor, followed by y and return.
chmod +x sequenceData/1-scripts/cutadaptParsing.py
nano sequenceData/1-scripts/example-script.sh
./sequenceData/1-scripts/cutadaptParsing.py sequenceData/0-metadata/cutadapt_primer_trimming.txt NTC_1_merged.fastq+NTC_2_merged.fastq+NTC_3_merged.fastq
Press ctrl + x and y to save the file and change the name to check_neg_trimming.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/check_neg_trimming.sh
Output
NTC_1_merged.fastq summary statistics:
=== Summary ===
Total reads processed: 306
Reads with adapters: 306 (100.0%)
Reverse-complemented: 0 (0.0%)
== Read fate breakdown ==
Reads that were too short: 77 (25.2%)
Reads that were too long: 3 (1.0%)
Reads discarded as untrimmed: 0 (0.0%)
Reads written (passing filters): 226 (73.9%)
Total basepairs processed: 88,071 bp
Total written (filtered): 70,668 bp (80.2%)
NTC_2_merged.fastq summary statistics:
=== Summary ===
Total reads processed: 7,842
Reads with adapters: 7,839 (100.0%)
Reverse-complemented: 0 (0.0%)
== Read fate breakdown ==
Reads that were too short: 7,691 (98.1%)
Reads that were too long: 6 (0.1%)
Reads discarded as untrimmed: 0 (0.0%)
Reads written (passing filters): 145 (1.8%)
Total basepairs processed: 507,546 bp
Total written (filtered): 45,343 bp (8.9%)
NTC_3_merged.fastq summary statistics:
=== Summary ===
Total reads processed: 6,114
Reads with adapters: 6,105 (99.9%)
Reverse-complemented: 0 (0.0%)
== Read fate breakdown ==
Reads that were too short: 5,928 (97.0%)
Reads that were too long: 7 (0.1%)
Reads discarded as untrimmed: 0 (0.0%)
Reads written (passing filters): 179 (2.9%)
Total basepairs processed: 440,273 bp
Total written (filtered): 55,985 bp (12.7%)
The cutadapt summary statistics for the negative control samples shows that a large proportion of the reads were discarded as they were too short (NTC_2: 98.1%; NTC_3: 97.0%). This probably meant that some primer-dimer sequences seeped through during the size selection step of the library preparation protocol and got sequenced. While primer-dimers should be removed during library preparation, as Illumina favours sequencing shorter fragments, such a low number as observed in this tutorial data set is a normal occurrence and nothing to be worried about.
4. Quality filtering#
During the bioinformatic pipeline, it is critical to only retain high-quality reads to reduce the abundance and impact of spurious sequences. There is an intrinsic error rate to all polymerases used during PCR amplification, as well as sequencing technologies. For example, the most frequently used polymerase during PCR is Taq, though lacks 3’ to 5’ exonuclease proofreading activity, resulting in relatively low replication fidelity. These errors will generate some portion of sequences that vary from their biological origin sequence. Such reads can substantially inflate metrics such as alpha diversity, especially in a denoising approach (more on this later). While it is near-impossible to remove all of these sequences bioinformatically, especially PCR errors, we will attempt to remove erroneous reads by filtering on base calling quality scores (the fourth line of a sequence record in a .fastq file).
Important
When processing your own metabarcoding data, I suggest you follow the same structure as we did with merging and trimming reads, i.e., check the code on one sample, play around with the parameter settings, and only batch process your samples when everything works fine. However, to save time during this tutorial, we will move straight to batch processing the samples for the quality filtering steps with the correct parameters.
4.1 Batch processing#
For quality filtering, we will be using the --fastq_filter command in VSEARCH. During this step, we will discard all sequences that do not adhere to a specific set of rules. Quality filtering parameters are not standardized, but rather specific for each library. For our tutorial data, we will filter out all sequences on a much stricter size range compared to the settings we used during primer trimming (--fastq_minlen 310 and --fastq_maxlen 316). Additionally, we will remove sequences that have ambiguous base calls, bases denotes as N, rather than A, C, G, or T (--fastq_maxns 0). Ambiguous base calls, however, are not a real issue with Illumina sequencing data. The last parameter we will use to filter our sequences is a maximum expected error rate (--fastq_maxee 1.0). As this is the last step in our bioinformatic pipeline where quality scores are essential, we will export our output files both in .fastq and .fasta format. Additionally, we will be merging all files together after this step in the bioinformatic pipeline. Before merging all files, however, we need to change the sequence headers to contain the information to which sample the sequence belongs to, which is currently stored in the file name. We can rename sequence header based on the file name using the --relabel parameter.
nano sequenceData/1-scripts/example-script.sh
cd sequenceData/4-demux/
for fq in *_trimmed.fastq
do
echo "Merging reads for: ${fq/_trimmed.fastq/}"
vsearch --fastq_filter ${fq} --fastq_maxee 1.0 --fastq_maxlen 316 --fastq_minlen 310 --fastq_maxns 0 --fastqout ../5-filter/${fq/_trimmed.fastq/_filtered.fastq} --fastaout ../5-filter/${fq/_trimmed.fastq/_filtered.fasta} --relabel ${fq/_trimmed.fastq/}.
done
Press ctrl + x and y to save the file and change the name to quality_filter.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/quality_filter.sh
Output
Merging reads for: COI_100_HK37
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
128093 sequences kept (of which 0 truncated), 8316 sequences discarded.
Merging reads for: COI_100_HK38
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
105809 sequences kept (of which 0 truncated), 6969 sequences discarded.
Merging reads for: COI_100_HK39
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
145327 sequences kept (of which 0 truncated), 9152 sequences discarded.
Merging reads for: COI_100_HK40
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
103665 sequences kept (of which 0 truncated), 6297 sequences discarded.
Merging reads for: COI_100_HK41
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
91076 sequences kept (of which 0 truncated), 5477 sequences discarded.
Merging reads for: COI_100_HK42
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
134371 sequences kept (of which 0 truncated), 8278 sequences discarded.
Merging reads for: COI_100_HK49
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
82338 sequences kept (of which 0 truncated), 5241 sequences discarded.
Merging reads for: COI_100_HK50
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
72895 sequences kept (of which 0 truncated), 4298 sequences discarded.
Merging reads for: COI_100_HK51
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
75547 sequences kept (of which 0 truncated), 5111 sequences discarded.
Merging reads for: COI_100_HK52
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
67509 sequences kept (of which 0 truncated), 3961 sequences discarded.
Merging reads for: COI_100_HK53
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
70090 sequences kept (of which 0 truncated), 4008 sequences discarded.
Merging reads for: COI_100_HK54
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
68948 sequences kept (of which 0 truncated), 3952 sequences discarded.
Merging reads for: COI_500_HK37
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
98678 sequences kept (of which 0 truncated), 6897 sequences discarded.
Merging reads for: COI_500_HK38
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
78856 sequences kept (of which 0 truncated), 4859 sequences discarded.
Merging reads for: COI_500_HK39
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
122200 sequences kept (of which 0 truncated), 7674 sequences discarded.
Merging reads for: COI_500_HK40
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
99565 sequences kept (of which 0 truncated), 6561 sequences discarded.
Merging reads for: COI_500_HK41
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
106658 sequences kept (of which 0 truncated), 6567 sequences discarded.
Merging reads for: COI_500_HK42
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
70167 sequences kept (of which 0 truncated), 4090 sequences discarded.
Merging reads for: COI_500_HK49
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
61051 sequences kept (of which 0 truncated), 4086 sequences discarded.
Merging reads for: COI_500_HK50
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
68680 sequences kept (of which 0 truncated), 4195 sequences discarded.
Merging reads for: COI_500_HK51
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
95654 sequences kept (of which 0 truncated), 6049 sequences discarded.
Merging reads for: COI_500_HK52
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
104924 sequences kept (of which 0 truncated), 4927 sequences discarded.
Merging reads for: COI_500_HK53
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
91320 sequences kept (of which 0 truncated), 5605 sequences discarded.
Merging reads for: COI_500_HK54
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
87233 sequences kept (of which 0 truncated), 4500 sequences discarded.
Merging reads for: NTC_1
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
161 sequences kept (of which 0 truncated), 65 sequences discarded.
Merging reads for: NTC_2
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
86 sequences kept (of which 0 truncated), 59 sequences discarded.
Merging reads for: NTC_3
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading input file 100%
92 sequences kept (of which 0 truncated), 87 sequences discarded.
Exercise 3
How would you check if the sequence headers of the .fastq and .fasta output files are relabeled and contain the sample information?
Answer 3
Using the head command, we can print the first N lines of a document.
head -n 1 sequenceData/4-demux/COI_100_HK37_trimmed.fastq
head -n 1 sequenceData/5-filter/COI_100_HK37_filtered.fastq
head -n 1 sequenceData/5-filter/COI_100_HK37_filtered.fasta
Using the tail command we can print the last N lines of a document. Note the different number of lines needed to be printed for the .fasta file.
tail -n 4 sequenceData/4-demux/COI_100_HK37_trimmed.fastq
tail -n 4 sequenceData/5-filter/COI_100_HK37_filtered.fastq
tail -n 5 sequenceData/5-filter/COI_100_HK37_filtered.fasta
Using the grep command on the sample name within the file, we can verify its presence in the file and also count the number of lines (sequences) that contain the sample information. Note the different search term for the .fasta file, due to the different sequence header structure. The grep output for the filtered .fastq and .fasta files should be identical.
grep -c "^@COI_100_HK37" sequenceData/4-demux/COI_100_HK37_trimmed.fastq
grep -c "^@COI_100_HK37" sequenceData/5-filter/COI_100_HK37_filtered.fastq
grep -c "^>COI_100_HK37" sequenceData/5-filter/COI_100_HK37_filtered.fasta
4.2 Summary output#
While VSEARCH provides the numbers for each sample before and after quality filtering, we will update the python script that generates the bar graph again, now to also include the filtered reads. This new python script takes in four user arguments, the same three as before, plus the path to where the filtered sequence files are stored. This will allow us to easily identify how the quality filtering performed and compare differences between samples.
nano sequenceData/1-scripts/rawMergedTrimmedFilteredStatistics.py
#! /usr/bin/env python3
## import modules
import os
import sys
import matplotlib.pyplot as plt
import pandas as pd
import seaborn as sns
## user arguments
mergedPath = sys.argv[1]
rawPath = sys.argv[2]
trimmedPath = sys.argv[3]
filteredPath = sys.argv[4]
## first, create a sample name list
mergedFileList = os.listdir(mergedPath)
sampleNameList = []
for mergedFile in mergedFileList:
sampleName = mergedFile.split('_merged.fastq')[0]
if sampleName.startswith('COI_') or sampleName.startswith('NTC_'):
sampleNameList.append(sampleName)
## count number of raw and merged sequences for each sample in sampleNameList
rawSeqCount = {}
mergedSeqCount = {}
trimmedSeqCount = {}
filteredSeqCount = {}
for sample in sampleNameList:
with open(f'{mergedPath}{sample}_merged.fastq', 'r') as mergedFile:
x = len(mergedFile.readlines()) / 4
mergedSeqCount[sample] = int(x)
with open(f'{rawPath}{sample}_1.fastq', 'r') as rawFile:
y = len(rawFile.readlines()) / 4
rawSeqCount[sample] = int(y)
with open(f'{trimmedPath}{sample}_trimmed.fastq', 'r') as trimmedFile:
z = len(trimmedFile.readlines()) / 4
trimmedSeqCount[sample] = int(z)
with open(f'{filteredPath}{sample}_filtered.fastq', 'r') as filteredFile:
a = len(filteredFile.readlines()) / 4
filteredSeqCount[sample] = int(a)
## create a dataframe from the dictionaries
df = pd.DataFrame({'Sample': list(rawSeqCount.keys()), 'Raw': list(rawSeqCount.values()), 'Merged': list(mergedSeqCount.values()), 'Trimmed': list(trimmedSeqCount.values()), 'Filtered': list(filteredSeqCount.values())})
## sort the dataframe by raw reads in descending order
df = df.sort_values(by='Raw', ascending=False)
## calculate the percentage of merged/raw and format it with 2 decimal places and the '%' symbol
df['Percentage'] = (df['Filtered'] / df['Raw'] * 100).round(2).astype(str) + '%'
## create a horizontal bar plot using seaborn
plt.figure(figsize=(20, 8)) # Adjust the figure size as needed
## use seaborn's barplot
ax = sns.barplot(x='Raw', y='Sample', data=df, label='Raw', color='#BBC6C8')
sns.barplot(x='Merged', y='Sample', data=df, label='Merged', color='#469597')
sns.barplot(x='Trimmed', y='Sample', data=df, label='Trimmed', color='#DDBEAA')
sns.barplot(x='Filtered', y='Sample', data=df, label='Filtered', color='#806491')
## add labels and title
plt.xlabel('Number of sequences')
plt.ylabel('Samples')
plt.title('Horizontal bar graph of raw, merged, trimmed, and filtered reads (Sorted by Total in Reverse)')
## add a legend
plt.legend()
# Add raw read count next to the bars
for i, v in enumerate(df['Percentage']):
ax.text(df['Raw'].values[i] + 50, i, v, va='center', fontsize=10, color='black')
## save the plot
plt.tight_layout()
plt.savefig('raw_and_merged_and_trimmed_and_filtered_bargraph.png', dpi = 300)
Press ctrl +x to exit out of the editor, followed by y and return.
chmod +x sequenceData/1-scripts/rawMergedTrimmedFilteredStatistics.py
nano sequenceData/1-scripts/example-script.sh
./sequenceData/1-scripts/rawMergedTrimmedFilteredStatistics.py sequenceData/2-raw/ sequenceData/2-raw/unzipped/ sequenceData/4-demux/ sequenceData/5-filter/
Press ctrl + x and y to save the file and change the name to check_quality_filter.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/check_quality_filter.sh
scp -r ednaw01@hpc2021-io1.hku.hk:~/ednaw01/raw_and_merged_and_trimmed_and_filtered_bargraph.png ./
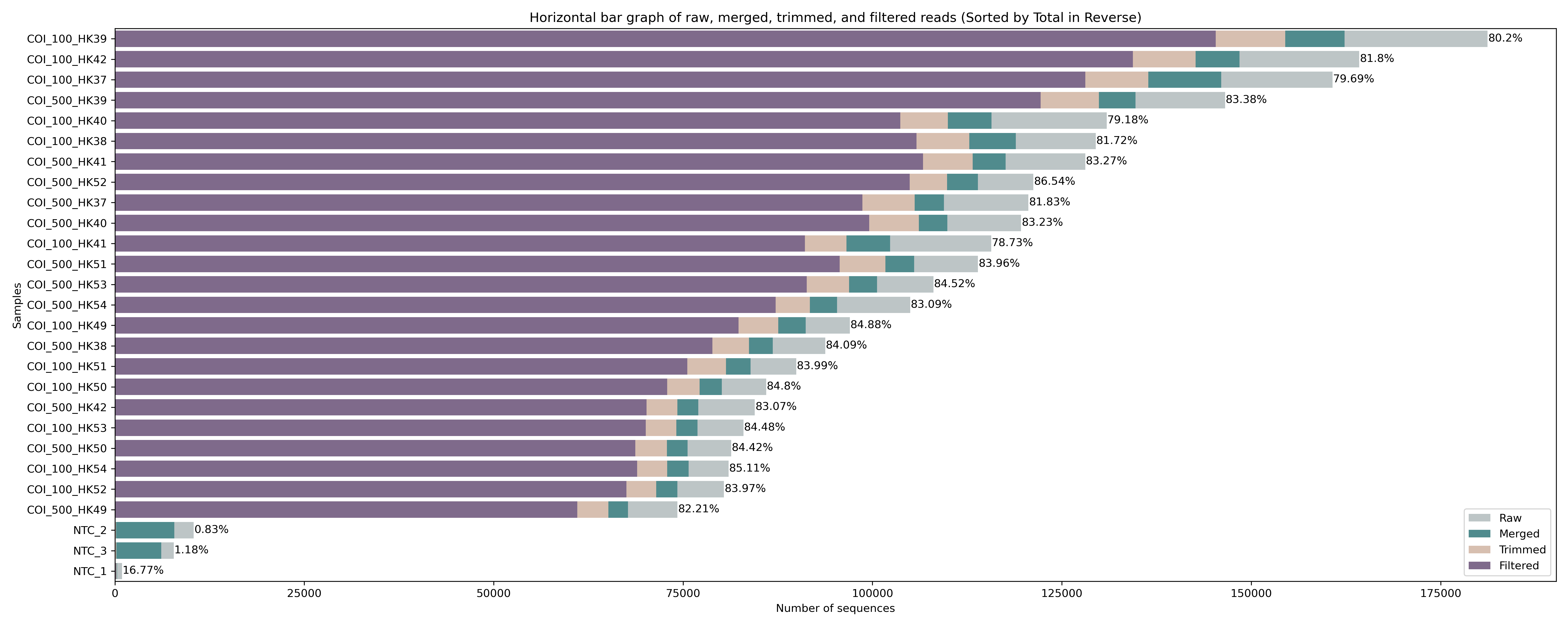
Fig. 13 : Read count of raw, merged, trimmed, and filtered files.#
This bar graph shows that after quality filtering, we still keep around 80% of the raw reads, which is very good and due to the high quality data we started with (remember the multiQC output from before!). Again, we see a big difference between the actual samples and negative controls is observed.
4.3 Check filtering step#
Before continuing with the bioinformatic pipeline, it is best to check if the quality filtering step retained only high-quality sequences that are of length ~313 bp (estimated length of the amplicon). If quality filtering was successful, we can move on to the next steps in our bioinformatic pipeline. The check the quality and length of our filtered sequence files, we will use FASTQC and MULTIQC, same as before.
nano sequenceData/1-scripts/example-script.sh
fastqc sequenceData/5-filter/*.fastq -o sequenceData/3-fastqc/ -t 8
Press ctrl + x and y to save the file and change the name to fastqc_quality_filter.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/fastqc_quality_filter.sh
Output
Started analysis of COI_100_HK37_filtered.fastq
Approx 5% complete for COI_100_HK37_filtered.fastq
Approx 10% complete for COI_100_HK37_filtered.fastq
Approx 15% complete for COI_100_HK37_filtered.fastq
Approx 20% complete for COI_100_HK37_filtered.fastq
Approx 25% complete for COI_100_HK37_filtered.fastq
Started analysis of COI_100_HK38_filtered.fastq
Approx 30% complete for COI_100_HK37_filtered.fastq
Approx 5% complete for COI_100_HK38_filtered.fastq
Approx 35% complete for COI_100_HK37_filtered.fastq
Approx 10% complete for COI_100_HK38_filtered.fastq
Approx 40% complete for COI_100_HK37_filtered.fastq
Approx 15% complete for COI_100_HK38_filtered.fastq
Approx 45% complete for COI_100_HK37_filtered.fastq
Approx 20% complete for COI_100_HK38_filtered.fastq
Approx 50% complete for COI_100_HK37_filtered.fastq
Approx 25% complete for COI_100_HK38_filtered.fastq
Approx 30% complete for COI_100_HK38_filtered.fastq
Approx 55% complete for COI_100_HK37_filtered.fastq
Approx 35% complete for COI_100_HK38_filtered.fastq
Approx 60% complete for COI_100_HK37_filtered.fastq
Approx 40% complete for COI_100_HK38_filtered.fastq
Approx 65% complete for COI_100_HK37_filtered.fastq
Approx 45% complete for COI_100_HK38_filtered.fastq
Approx 70% complete for COI_100_HK37_filtered.fastq
Approx 50% complete for COI_100_HK38_filtered.fastq
...
FastQC will generate a .html report for every single .fastq file in the subdirectory sequenceData/5-filter/. Since we are working with 27 files, it will be easier to compare the reports by collating them, something we can do using multiQC.
nano sequenceData/1-scripts/example-script.sh
multiqc sequenceData/3-fastqc/*filtered* -o sequenceData/3-fastqc/
Press ctrl + x and y to save the file and change the name to multiqc_quality_filter.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/multiqc_quality_filter.sh
scp -r ednaw01@hpc2021-io1.hku.hk:~/ednaw01/sequenceData/3-fastqc/multiqc_report.html ./
The multiQC program will combine all 27 FastQC reports into a single .html document. Let’s open this to see how our filtered sequence data is looking like. Since we have written both multiQC reports (raw and filtered) to the same sequenceData/3-fastqc/ directory, make sure to open the latest report. The file name will most likely end in _1.html.
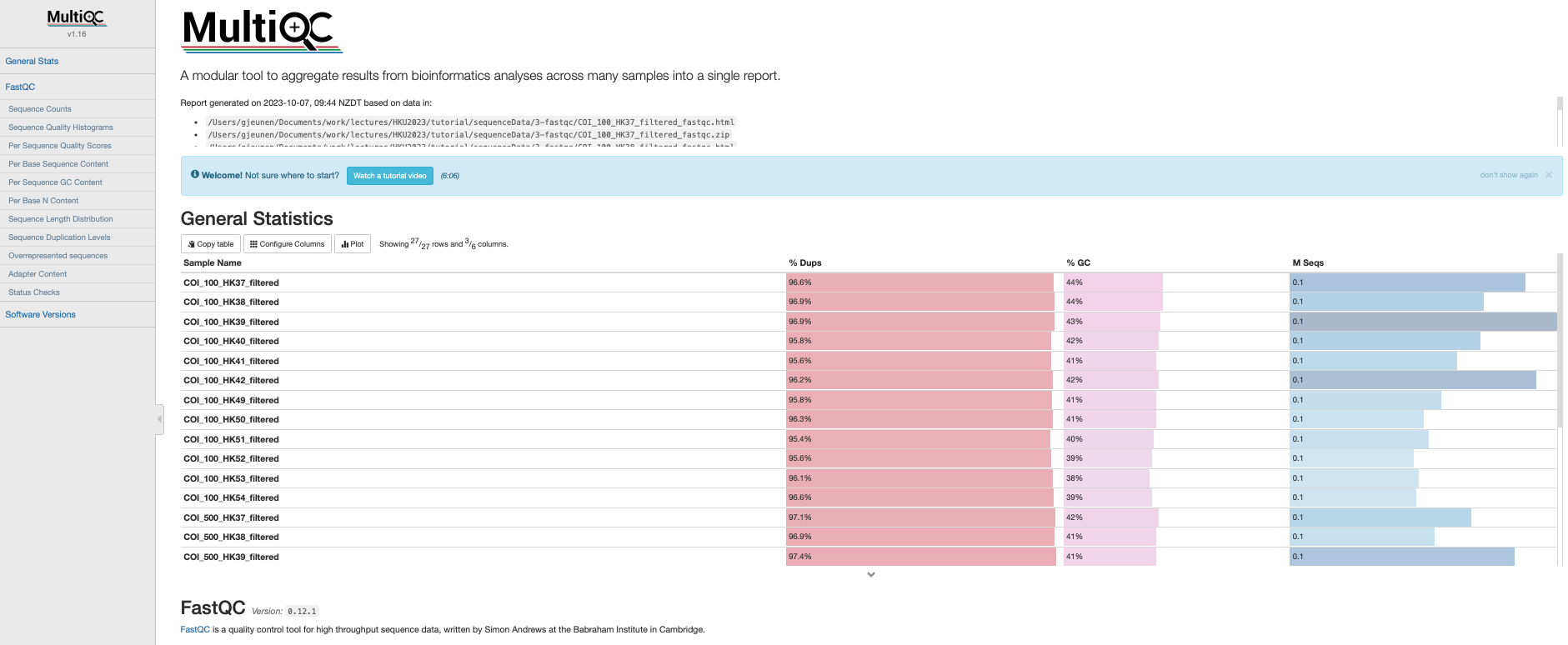
Fig. 14 : The .html multiQC report for the quality filtered reads#
From this multiQC report, we can see that we only retained high quality reads that are of length 313 bp. Exactly what we need to move forward with our bioinformatic pipeline!
5. Dereplication#
5.1 Combining data into one file#
Once we have verified that the reads passing quality filtering are of high quality and of a length similar to the expected amplicon size, we can move forward with our bioinformatic pipeline. For the next final steps of the bioinformatic pipeline, it makes more sense to combine all the files into a single file. So, let’s do this first using the cat command, which stands for concatenate. Since base calling quality scores are not essential for our analysis from this point onwards, we can combine all the .fasta files to reduce the file size and computational cost. To concatenate all files, we can use the * symbol. Make sure to specify the output file using >, otherwise cat will print the combined file to the Terminal window instead.
cat sequenceData/5-filter/*.fasta > sequenceData/6-quality/combined.fasta
Exercise 4
How would you determine if no sequences were lost during the cat command to merge all .fasta files?
Answer 4
First, let’s start with the easiest problem, i.e., determine the number of sequences in the merged file. Here we can simply pipe the output of wc -l to the awk command and divide the number of lines by 5 (= one sequence record).
wc -l sequenceData/6-quality/combined.fasta | awk '{print $1/5}'
Output
2230993
To count the total number of sequences in the separate files before concatenation, we will need to write a for loop to count all the lines in each file, pipe that number to awk to divide it by 5, and sum the number of sequences for each file.
total_lines=0
for item in sequenceData/5-filter/*.fasta
do
lines=$(wc -l "$item" | awk '{print $1/5}')
total_lines=$((total_lines + lines))
done
echo "$total_lines"
Output
2230993
5.2 Finding unique sequences#
With the cat command, we have created a single file. The next step in our bioinformatic pipeline is to dereplicate our data, i.e., find unique sequences. Since metabarcoding data is based on a PCR amplification method, the same DNA molecule will have been copied thousands or millions of times and, therefore, sequenced multiple times. In order to reduce the file size and computational cost, it is convenient to work with unique sequences and keep track of how many times each unique sequence was found in our data set. This dereplication step is also the reason why we combined all samples into a single file, as the same barcode (or species if you will) can be found across different samples.
We can dereplicate our data by using the --derep_fulllength command in VSEARCH. The --sizeout parameter keeps a tally of the number of times each unique sequence was observed across all samples and places this information in the sequence header. Since we are looking at unique sequences across samples, it wouldn’t make much sense to keep the current sequence header information, as it currently refers to which sample a sequence belongs to. We will, therefore, rename the sequence headers by using the --relabel parameter.
nano sequenceData/1-scripts/example-script.sh
vsearch --derep_fulllength sequenceData/6-quality/combined.fasta --sizeout --relabel uniq. --output sequenceData/6-quality/uniques.fasta
Press ctrl + x and y to save the file and change the name to dereplication.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/dereplication.sh
Output
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Dereplicating file sequenceData/6-quality/combined.fasta 100%
697630765 nt in 2230993 seqs, min 310, max 316, avg 313
Sorting 100%
244379 unique sequences, avg cluster 9.1, median 1, max 68022
Writing FASTA output file 100%
We can see from the VSEARCH output that 244,379 out of 2,230,993 sequences were unique. The average number a unique sequence was observed was 9.1 times, while the median is 1 and the maximum is 68,022 times. As mentioned before, the --sizeout parameter will have added the abundance information for each unique sequence to the header. Let’s use the head command to investigate the first couple of sequences.
head -n 10 sequenceData/6-quality/uniques.fasta
Output
>uniq.1;size=68022
CCTAAGCTCCAATATTGCCCACGCCGGGGCGTCTGTTGACCTTGCTATCTTTAGGCTACACTTGGCTGGGGTTTCTTCTC
TACTCGGGGCTGTAAACTTTATTAGAACTATCGCTAACCTGCGAGCTTTAGGGCTAATTCTTGACCGTATAACACTATTC
ACATGATCAGTTCTTATCACCGCCATCCTTCTCCTTCTTTCTCTACCTGTTCTCGCAGGGGCTATTACGATACTCCTTAC
CGACCGAAATCTAAATACCTCTTTTTATGACCCTAGAGGAGGGGGAGACCCTATTCTCTACCAACACCTGTTT
>uniq.2;size=59045
CCTCTCCGCTAGCGGAGCCCATGGATCTGCCTCAGTAGATTTAAGGATCTTTTCCCTCCATCTAGCAGGAGTATCTTCTA
TCCTCGGATCCATCAACTTTATCACCACAATTATTAATATGCGAACCCCAAACCTCACATGAGACAAAATCTCTCTCTTT
ACTTGATCCATCCTTATTACTACTATCCTACTCCTCCTCTCTCTCCCTGTCCTAGCCGGAGCTTTAACTATACTTCTAAC
AGACCGTAACTTCAACACAACATTTTTTGACCCTAGAGGAGGAGGCGACCCTATCCTTTACCAGCACCTTTTC
The output of the head command shows that the unique sequences are already sorted by abundance, with the most abundant unique sequence occurring 68,022 times and the second most abundant sequence occurring 59,045 times.
Exercise 5
How would you determine the number of times the 10 most and 10 least abundant unique sequences were observed?
Answer 5
The simplest way to determine the abundance of the 10 most and 10 least abundant unique sequences, based on the code we’ve learned during this tutorial thus far, would be to write two lines of code, one for the 10 most abundant and one for the 10 least abundant. We can solve this problem by taking out all the lines that start with ^>uniq using grep and pipe that list to either the head -n 10 or tail -n 10 command.
grep '^>uniq' sequenceData/6-quality/uniques.fasta | head -n 10
grep '^>uniq' sequenceData/6-quality/uniques.fasta | tail -n 10
Output
>uniq.1;size=68022
>uniq.2;size=59045
>uniq.3;size=57634
>uniq.4;size=42670
>uniq.5;size=42507
>uniq.6;size=40947
>uniq.7;size=38849
>uniq.8;size=38764
>uniq.9;size=37125
>uniq.10;size=34995
>uniq.244370;size=1
>uniq.244371;size=1
>uniq.244372;size=1
>uniq.244373;size=1
>uniq.244374;size=1
>uniq.244375;size=1
>uniq.244376;size=1
>uniq.244377;size=1
>uniq.244378;size=1
>uniq.244379;size=1
To solve this problem with one line of code, we need to use awk. As mentioned before, the syntax of this language is quite complex. To provide a bit of background to the solution:
/^>uniq/: is the pattern we are looking for (similar togrep).{a[i++]=$0}: when a line matches the pattern, it is added to an arraya, and the indexiis incremented.$0represents the entire line.END: the next section of the code is only executed after processing all lines in the input file.for (j=i-10; j<i; j++) print a[j]: this loop prints the last 10 lines that matched the pattern. It starts fromi - 10toi, whereiis the number of lines that matched the pattern, and prints those lines from the arraya.for (k=0; k<10; k++) print a[k]: this loop prints the first 10 lines that matched the pattern, iterating from0to9(first 10 elements) and prints those lines from the arraya.
awk '/^>uniq/ {a[i++]=$0} END {for (j=i-10; j<i; j++) print a[j]; for (k=0; k<10; k++) print a[k]}' sequenceData/6-quality/uniques.fasta
Output
>uniq.244370;size=1
>uniq.244371;size=1
>uniq.244372;size=1
>uniq.244373;size=1
>uniq.244374;size=1
>uniq.244375;size=1
>uniq.244376;size=1
>uniq.244377;size=1
>uniq.244378;size=1
>uniq.244379;size=1
>uniq.1;size=68022
>uniq.2;size=59045
>uniq.3;size=57634
>uniq.4;size=42670
>uniq.5;size=42507
>uniq.6;size=40947
>uniq.7;size=38849
>uniq.8;size=38764
>uniq.9;size=37125
>uniq.10;size=34995
6. Denoising#
Now that our data set is filtered and unique sequences have been retrieved, we are ready for the next step in the bioinformatic pipeline, i.e., looking for biologically meaningful or biologically correct sequences. Two approaches to achieve this goal exist, including denoising and clustering. There is still an ongoing debate on what the best approach is to obtain these biologicall meaningful sequences. For more information, these are two good papers to start with: Brandt et al., 2021 and Antich et al., 2021. For this tutorial we won’t be discussing this topic in much detail, but this is the basic idea…
When clustering the dataset, OTUs (Operational Taxonomic Units) will be generated by combining sequences that are similar to a set percentage level (traditionally 97%), with the most abundant sequence identified as the true sequence. When clustering at 97%, this means that if a sequence is more than 3% different than the generated OTU, a second OTU will be generated. The concept of OTU clustering was introduced in the 1960s and has been debated since. Clustering the dataset is usually used for identifying species in metabarcoding data.
Denoising, on the other hand, attempts to identify all correct biological sequences through an algorithm. In short, denoising will cluster the data with a 100% threshold and tries to identify errors based on abundance differences. The retained sequences are called ZOTU (Zero-radius Operation Taxonomic Unit) or ASVs (Amplicon Sequence Variants). Denoising the dataset is usually used for identifying intraspecific variation in metabarcoding data. A schematic of both approaches can be found below.
Fig. 15 : A schematic representation of denoising and clustering. Copyright: Antich et al., 2021#
This difference in approach may seem small but has a very big impact on your final dataset!
When you denoise the dataset, it is expected that one species may have more than one ZOTU. This means that when the reference database is incomplete, or you plan to work with ZOTUs instead of taxonomic assignments, your diversity estimates will be highly inflated. When clustering the dataset, on the other hand, it is expected that an OTU may have more than one species assigned to it, meaning that you may lose some correct biological sequences that are present in your data by merging species with barcodes more similar than 97%. In other words, you will miss out on differentiating closely related species and intraspecific variation.
For this tutorial, we will use the denoising approach, as it is favoured in recent years. However, clustering is still a valid option. So, feel free to explore this approach for your own data set if you prefer.
For denoising, we will use the unoise3 algorithm as implemented in the --cluster_unoise command in VSEARCH. Since denoising is based on read abundance of the unique sequences, we can specify the --sizein parameter. The minimum abundance threshold for a true denoised read is defaulted to 8 reads, as specified by the unoise3 algorithm developer. However, more recent research by Bokulich et al., 2013, identified a minimum abundance threshold to be more appropriate. Hence, we will set the --minsize parameter to 0.001%, which in our case is ~22 reads. As we will be merging different reads with varying abundances, we need to recalculate the new count for each denoised read using the --sizeout parameter. Relabeling the sequence headers can be done through the --relabel parameter and the output file is specified using --centroids.
nano sequenceData/1-scripts/example-script.sh
vsearch --cluster_unoise sequenceData/6-quality/uniques.fasta --sizein --minsize 22 --sizeout --relabel denoised. --centroids sequenceData/6-quality/denoised.fasta
Press ctrl + x and y to save the file and change the name to denoising.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/denoising.sh
Output
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading file sequenceData/6-quality/uniques.fasta 100%
1519235 nt in 4859 seqs, min 310, max 316, avg 313
minsize 22: 239520 sequences discarded.
Masking 100%
Sorting by abundance 100%
Counting k-mers 100%
Clustering 100%
Sorting clusters 100%
Writing clusters 100%
Clusters: 1372 Size min 22, max 187320, avg 3.5
Singletons: 0, 0.0% of seqs, 0.0% of clusters
From the output, we can see that we have generated 1,372 denoised reads and discarded 239,520 unique sequences, as they did not achieve an abundance of at least 22 reads.
7. Chimera removal#
The second to last step in our bioinformatic pipeline is to remove chimeric sequences. Amplicon sequencing has the potential to generate chimeric reads, which can cause spurious inference of biological variation. Chimeric amplicons form when an incomplete DNA strand anneals to a different template and primes synthesis of a new template derived from two different biological sequences, or in other words chimeras are artefact sequences formed by two or more biological sequences incorrectly joined together. More information can be found on this website and a simple illustration can be found below.
Fig. 16 : A schematic representation of chimera formation. Copyright: Genome Research#
We will use the --uchime3_denovo algorithm as implemented in VSEARCH for removing chimeric sequences from our denoised reads. This method is also based on sequence abundance, hence, we need to provide the --sizein parameter. As the output (parameter --nonchimeras) file will contain our biologically relevant sequences, we will relabel our sequence headers using --relabel zotu..
nano sequenceData/1-scripts/example-script.sh
vsearch --uchime3_denovo sequenceData/6-quality/denoised.fasta --sizein --nonchimeras sequenceData/8-final/zotus.fasta --relabel zotu.
Press ctrl + x and y to save the file and change the name to chimeras.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/chimeras.sh
Output
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading file sequenceData/6-quality/denoised.fasta 100%
429255 nt in 1372 seqs, min 310, max 316, avg 313
Masking 100%
Sorting by abundance 100%
Counting k-mers 100%
Detecting chimeras 100%
Found 27 (2.0%) chimeras, 1345 (98.0%) non-chimeras,
and 0 (0.0%) borderline sequences in 1372 unique sequences.
Taking abundance information into account, this corresponds to
1085 (0.1%) chimeras, 1785492 (99.9%) non-chimeras,
and 0 (0.0%) borderline sequences in 1786577 total sequences.
The output shows that 98% of our denoised reads were kept and 27 sequences were identified as chimeric. Taking into account abundance information, this would result into 0.1% of reads identified as chimeras.
The sequenceData/8-final/zotus.fasta file is the first output file we have created that we need for our statistical analysis!
Important
If you use the unoise3 algorithm as implemented in USEARCH, chimera removal is built into the denoising step and will not have to be conducted separately!
8. Frequency table#
Now that we have created our list of biologically relevant sequences or ZOTUs or “species”, we are ready to generate a frequency table, also known as a count table. This will be the last step in our bioinformatic pipeline. A frequency table is something you might have encountered before during some traditional ecological surveys you have conducted, whereby a table was created with site names as column headers, a species list as rows, and the number in each cell representing the number of individuals observed for a specific species at a specific site.
In metabarcoding studies, a frequency table is analogous, where it tells us how many times each sequence has appeared in each sample. It is the end-product of all the bioinformatic processing steps we have conducted today. Now that we have identified what we believe to be true biological sequences, we are going to generate our frequency table by matching the merged sequences to our ZOTU sequence list. Remember that the sequences within the sequenceData/6-quality/combined.fasta file have the information of the sample they belong to present in the sequence header, which is how the --usearch_global command in VSEARCH can generate the frequency table. The --db parameter allows us to set the ZOTU sequence list (sequenceData/8-final/zotus.fasta) as the database to search against, while we can specify the --strand parameter as plus, since all sequences are in the same direction after primer trimming. Finally, we need to incorporate a 97% identity threshold for this function through the --id parameter. This might seem counter-intuitive, since we employed a denoising approach. However, providing some leniency on which sequences can map to our ZOTU will allow us to incorporate a larger percentage of the data set. As some ZOTUs might be more similar to each other than 97%, the algorithm will sort out the best match and add the sequence to the correct ZOTU sequence. If you’d like to be more conservative, you can set this threshold to 99%, though this is not recommended by the authors.
nano sequenceData/1-scripts/example-script.sh
vsearch --usearch_global sequenceData/6-quality/combined.fasta --db sequenceData/8-final/zotus.fasta --strand plus --id 0.97 --otutabout sequenceData/8-final/zotutable.txt
Press ctrl + x and y to save the file and change the name to frequency_table.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/frequency_table.sh
Output
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading file sequenceData/8-final/zotus.fasta 100%
420804 nt in 1345 seqs, min 310, max 316, avg 313
Masking 100%
Counting k-mers 100%
Creating k-mer index 100%
Searching 100%
Matching unique query sequences: 2195816 of 2230993 (98.42%)
Writing OTU table (classic) 100%
The output printed to the Terminal window shows we managed to map 2,195,816 (98.42%) sequences to the ZOTU list, which were incorporated in our frequency table. The output file is a simple text file, which we can open in Excel. Below is a screenshot where I coloured the samples (column headers) blue and the sequence list (row names) yellow.
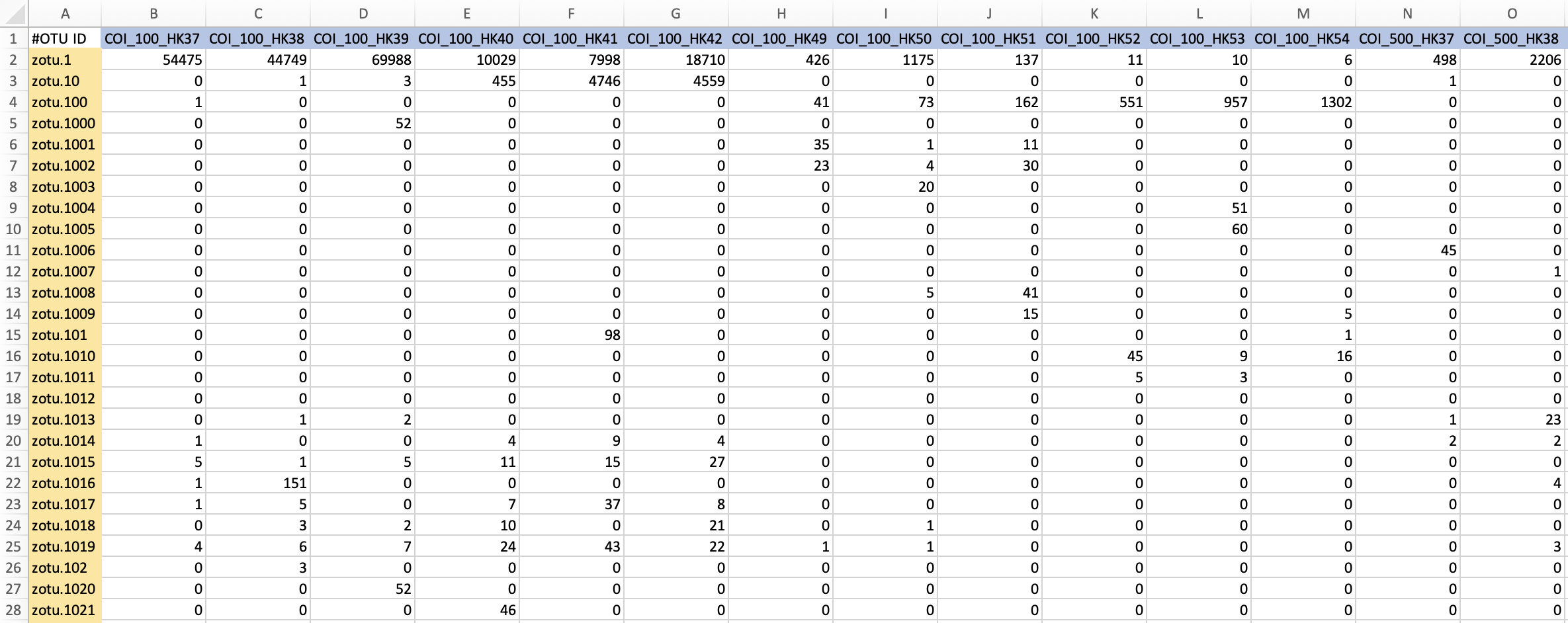
Fig. 17 : The frequency table output file in Excel, where the samples are coloured blue and the sequences are coloured yellow.#
8.1 Summarise sequence counts#
As a final step, let’s update the python script that produces a bar graph again, now including the final read count incorporated for each sample. This new python script takes in five user arguments, the same four as before, plus the frequency table file name. This final bar graph will allow us to get a great overview of what we have accomplished today.
nano sequenceData/1-scripts/rawMergedTrimmedFilteredFinalStatistics.py
#! /usr/bin/env python3
## import modules
import os
import sys
import matplotlib.pyplot as plt
import pandas as pd
import seaborn as sns
## user arguments
mergedPath = sys.argv[1]
rawPath = sys.argv[2]
trimmedPath = sys.argv[3]
filteredPath = sys.argv[4]
freqTable = sys.argv[5]
## first, create a sample name list
mergedFileList = os.listdir(mergedPath)
sampleNameList = []
for mergedFile in mergedFileList:
sampleName = mergedFile.split('_merged.fastq')[0]
if sampleName.startswith('COI_') or sampleName.startswith('NTC_'):
sampleNameList.append(sampleName)
## count number of raw and merged sequences for each sample in sampleNameList
rawSeqCount = {}
mergedSeqCount = {}
trimmedSeqCount = {}
filteredSeqCount = {}
for sample in sampleNameList:
with open(f'{mergedPath}{sample}_merged.fastq', 'r') as mergedFile:
x = len(mergedFile.readlines()) / 4
mergedSeqCount[sample] = int(x)
with open(f'{rawPath}{sample}_1.fastq', 'r') as rawFile:
y = len(rawFile.readlines()) / 4
rawSeqCount[sample] = int(y)
with open(f'{trimmedPath}{sample}_trimmed.fastq', 'r') as trimmedFile:
z = len(trimmedFile.readlines()) / 4
trimmedSeqCount[sample] = int(z)
with open(f'{filteredPath}{sample}_filtered.fastq', 'r') as filteredFile:
a = len(filteredFile.readlines()) / 4
filteredSeqCount[sample] = int(a)
freqTableDF = pd.read_csv(freqTable, delimiter = '\t', index_col = 0).transpose()
freqTableDF['Final'] = freqTableDF.sum(axis = 1)
columnsToDrop = freqTableDF.columns.difference(['Final'])
freqTableDF.drop(columns = columnsToDrop, inplace = True)
## create a dataframe from the dictionaries
df = pd.DataFrame({'Sample': list(rawSeqCount.keys()), 'Raw': list(rawSeqCount.values()), 'Merged': list(mergedSeqCount.values()), 'Trimmed': list(trimmedSeqCount.values()), 'Filtered': list(filteredSeqCount.values())})
## merge both data frames
merged_df = df.merge(freqTableDF, left_on = ['Sample'], right_index = True, how = 'inner')
## sort the dataframe by raw reads in descending order
merged_df = merged_df.sort_values(by='Raw', ascending=False)
## calculate the percentage of merged/raw and format it with 2 decimal places and the '%' symbol
merged_df['Percentage'] = (merged_df['Final'] / merged_df['Raw'] * 100).round(2).astype(str) + '%'
## create a horizontal bar plot using seaborn
plt.figure(figsize=(20, 8)) # Adjust the figure size as needed
## use seaborn's barplot
ax = sns.barplot(x='Raw', y='Sample', data=merged_df, label='Raw', color='#BBC6C8')
sns.barplot(x='Merged', y='Sample', data=merged_df, label='Merged', color='#469597')
sns.barplot(x='Trimmed', y='Sample', data=merged_df, label='Trimmed', color='#DDBEAA')
sns.barplot(x='Filtered', y='Sample', data=merged_df, label='Filtered', color='#806491')
sns.barplot(x='Final', y='Sample', data=merged_df, label='Final', color='#2F70AF')
## add labels and title
plt.xlabel('Number of sequences')
plt.ylabel('Samples')
plt.title('Horizontal bar graph of raw, merged, trimmed, and filtered reads (Sorted by Total in Reverse)')
## add a legend
plt.legend()
# Add raw read count next to the bars
for i, v in enumerate(merged_df['Percentage']):
ax.text(merged_df['Raw'].values[i] + 50, i, v, va='center', fontsize=10, color='black')
## save the plot
plt.tight_layout()
plt.savefig('raw_and_merged_and_trimmed_and_filtered_and_final_bargraph.png', dpi = 300)
Press ctrl +x to exit out of the editor, followed by y and return.
chmod +x sequenceData/1-scripts/rawMergedTrimmedFilteredFinalStatistics.py
nano sequenceData/1-scripts/example-script.sh
./sequenceData/1-scripts/rawMergedTrimmedFilteredFinalStatistics.py sequenceData/2-raw/ sequenceData/2-raw/unzipped/ sequenceData/4-demux/ sequenceData/5-filter/ sequenceData/8-final/zotutable.txt
Press ctrl + x and y to save the file and change the name to check_table.sh. Press return and y to save under a different name.
sbatch sequenceData/1-scripts/check_table.sh
scp -r ednaw01@hpc2021-io1.hku.hk:~/ednaw01/raw_and_merged_and_trimmed_and_filtered_and_final_bargraph.png ./
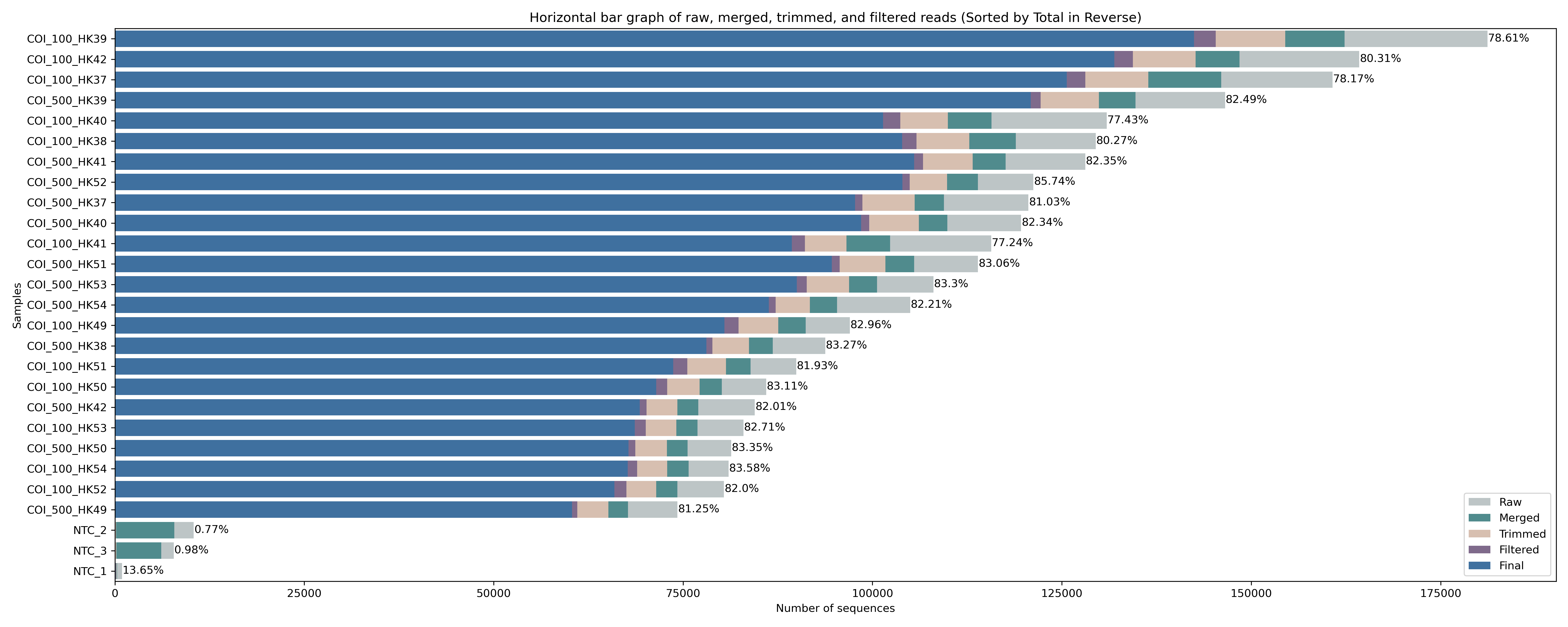
Fig. 18 : Read count of raw, merged, trimmed, filtered, and final frequency table for each sample.#
This bar graph shows that for the final frequency table, we have kept around 80% of the raw reads for each sample, which is very good and due to the high quality data we started with (remember the multiQC output from before!). Again, we see a big difference between the actual samples and negative controls is observed.
That’s it for today, see you tomorrow when we will assign a taxonomic ID to our ZOTU sequence list!