Taxonomy assignment#
1. Introduction#
As we have previously discussed, eDNA metabarcoding is the process whereby we amplify a specific gene region of the taxonomic group of interest from a DNA extract obtained from an environmental sample. In our tutorial data set, the COI sequence data originated from settlement plates that were placed in highly impacted and low-impact coastal regions in Hong Kong. After the bioinformatic processing we conducted in the previous section, we ended up with a frequency table (sequenceData/8-final/zotutable.txt) and a sequence file (sequenceData/8-final/zotus.fasta) containing all the biologically relevant sequences. The next step in the bioinformatic process will be to assign a taxonomic ID to each sequence to determine what species were detected through our eDNA metabarcoding analysis.
There are four basic strategies to taxonomy classification (and an endless number of variations of those four basic strategies):
sequence similarity (SS);
sequence composition (SC);
phylogenetic (Ph);
and probabilistic (Pr).
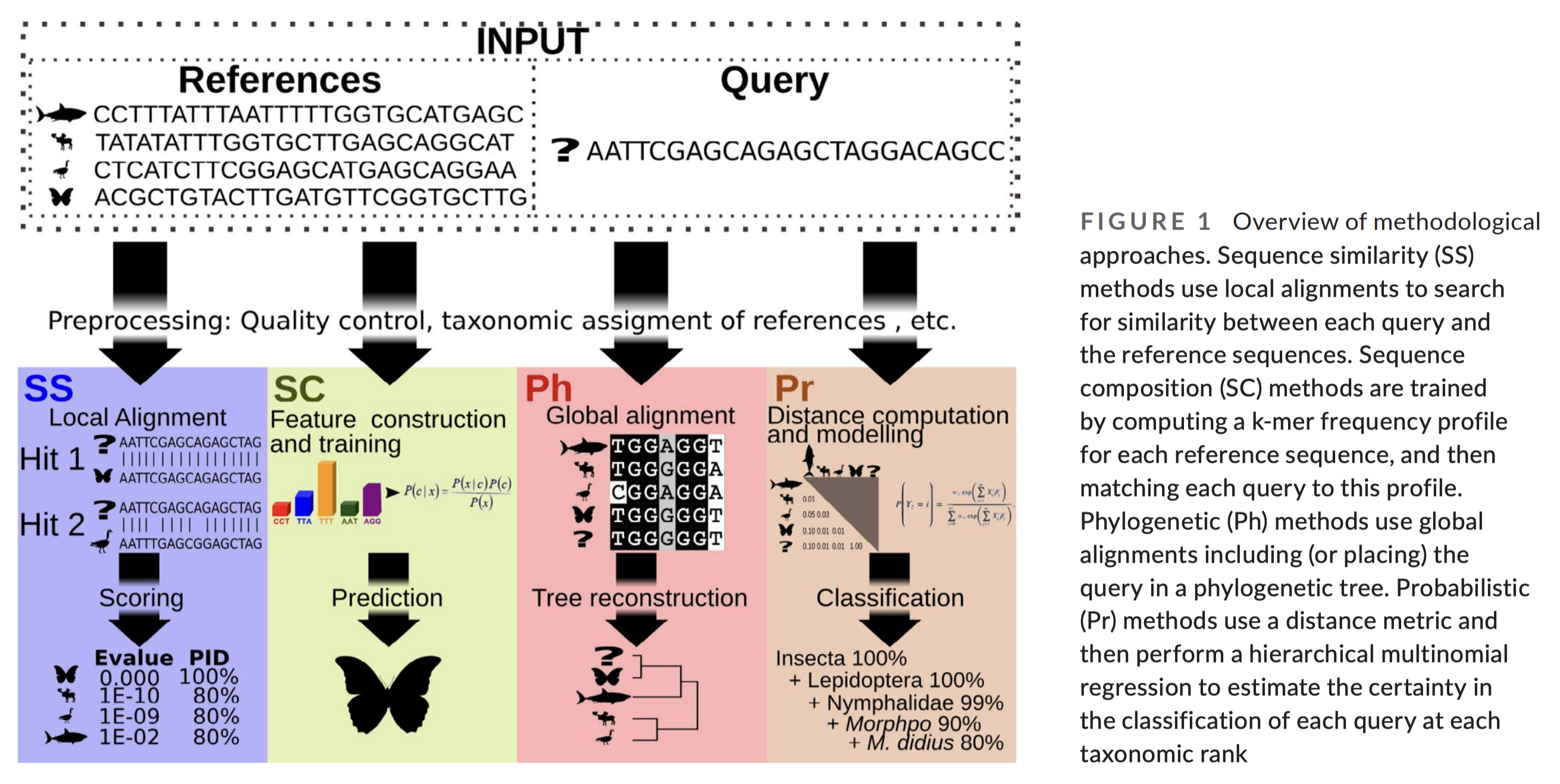
Fig. 19 : The four basic strategies to taxonomy classification. Copyright by Hleap et al., 2021.#
Of the four abovementioned strategies to taxonomy classification, the Sequency Similarity and Sequence Composition methods are currently the most frequently used in the metabarcoding research community. Multiple comparative experiments have been conducted to determine the most optimal approach to assign a taxonomic ID to metabarcoding data, though it seems to be that the main conclusion of those papers is that their own method is best. One example of a comparative study is the one from Bokulich et al., 2018.
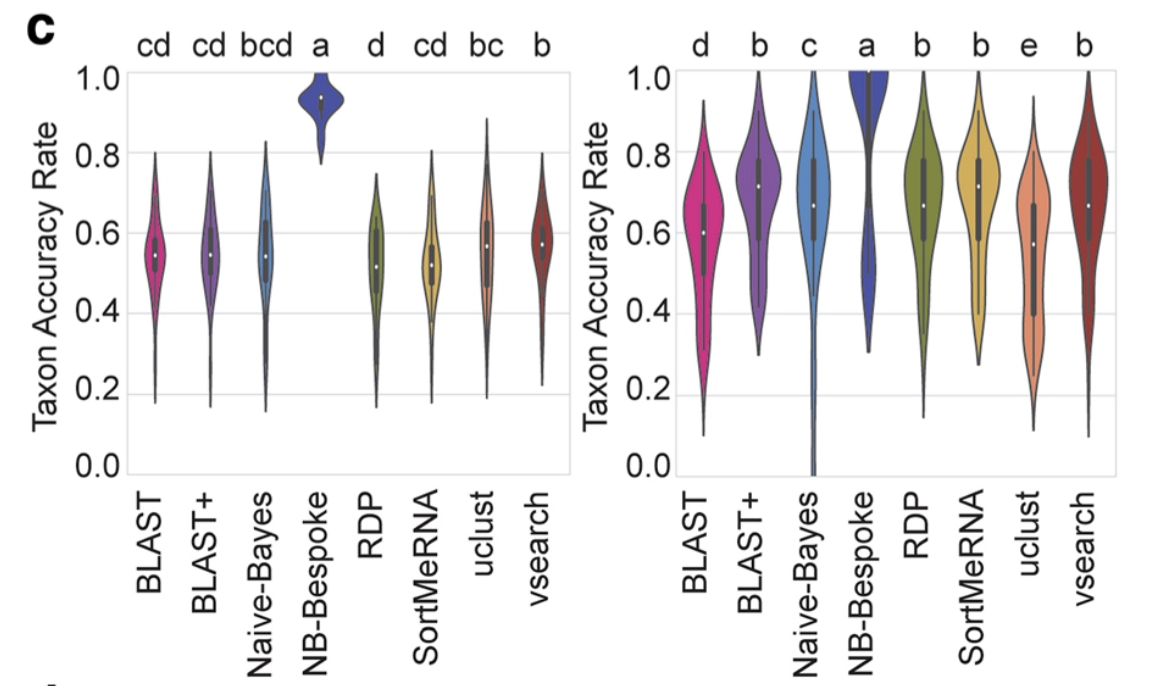
Fig. 20 : A comparison of taxonomy classifiers. Copyright by Bokulich et al., 2018.#
Within this tutorial, we will cover three different ways to assign a taxonomic ID to our sequences. However, before doing so, we will need to create a reference database that will be used by the classifiers to our ZOTU list to the reference barcodes.
2. Reference databases#
Several reference databases are available online. The most notable ones being NCBI, EMBL, BOLD, SILVA, and RDP. However, recent research has indicated a need to use custom curated reference databases to increase the accuracy of taxonomy assignment. While certain gene-specific or primer-specific reference databases are available (RDP: 16S microbial; MIDORI: COI eukaryotes; etc.), this essential data source is missing in most instances. We will, therefore, show you how to build your own custom curated reference database using the CRABS (Creating Reference databases for Amplicon-Based Sequencing) program. CRABS can be installed via GitHub, Docker, and conda.
Attention
Since the online COI reference material is quite large, it being the standard metazoan barcode gene, we will not build a COI reference database during this tutorial. Instead, we will use a pre-assembled COI database. However, to show you how to build your own database, we will go through the steps to build a 16S Chondrichthyes (sharks and rays) database, as the online reference material is small. Therefore, the steps can be accomplished quite quickly.
2.1 CRABS workflow#
The CRABS workflow consists of seven parts, including:
Downloading and importing data from multiple online repositories, including NCBI, BOLD, EMBL, MitoFish, etc.;
Retrieving amplicon regions through in silico PCR analysis;
Retrieving amplicons with missing primer-binding regions through Pairwise Global Alignments with amplicons from the in Silico PCR analysis as seed sequences;
Generating the taxonomic lineage for amplicons;
Curating the database via multiple filtering parameters;
Post-processing functions and visualizations to provide a summary overview of the final reference database; and
Exporting the custom curated reference database in various formats covering most format requirements of taxonomic assignment tools.
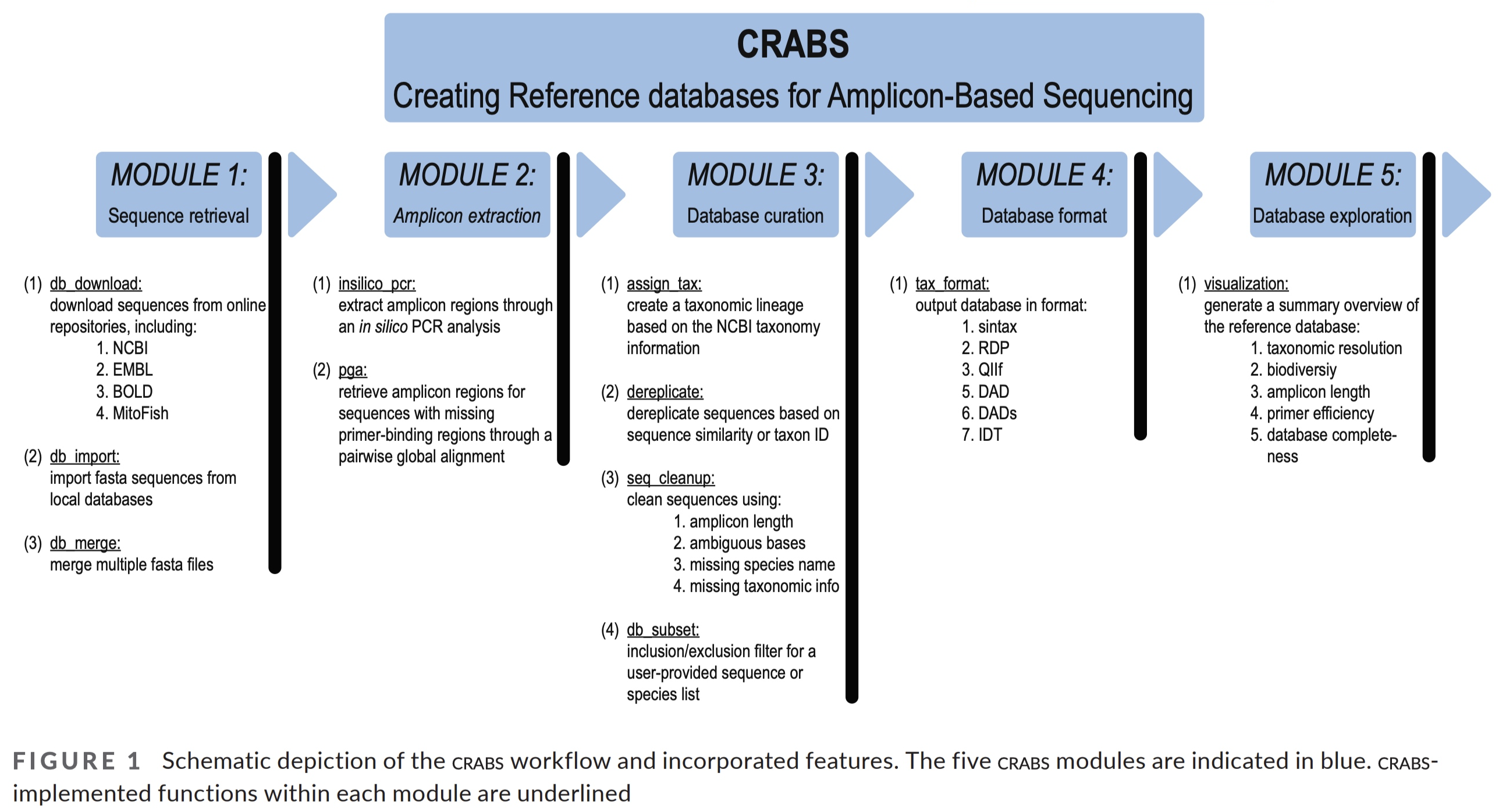
Fig. 21 : The CRABS worflow to create your own reference database. Copyright by Jeunen et al., 2022.#
2.2 Help documentation#
To bring up the general help documentation for CRABS, we can use the --help or -h command:
crabs --version
crabs --help
Output
(base) gjeunen@MacBook-Pro-16 tutorial % crabs --version
0.1.8
(base) gjeunen@MacBook-Pro-16 tutorial % crabs --help
usage: crabs [-h] [--version] {db_download,db_import,db_merge,insilico_pcr,pga,assign_tax,dereplicate,seq_cleanup,db_subset,visualization,tax_format} ...
creating a curated reference database
positional arguments:
{db_download,db_import,db_merge,insilico_pcr,pga,assign_tax,dereplicate,seq_cleanup,db_subset,visualization,tax_format}
options:
-h, --help show this help message and exit
--version show program's version number and exit
The output from this command shows the functions that are available within CRABS. To get a detailed overview and bring up the documentation of one of the functions, we can use the --help or -h parameter with the function.
crabs db_download -h
Output
usage: crabs db_download [-h] -s SOURCE [-db DATABASE] [-q QUERY] [-p SPECIES] [-m MARKER] [-o OUTPUT] [-k ORIG] [-d DISCARD] [-g BOLDGAP] [-e EMAIL]
[-b BATCHSIZE]
downloading sequence data from online databases
options:
-h, --help show this help message and exit
-s SOURCE, --source SOURCE
specify online database used to download sequences. Currently supported options are: (1) ncbi, (2) embl, (3) mitofish, (4) bold, (5)
taxonomy
-db DATABASE, --database DATABASE
specific database used to download sequences. Example NCBI: nucleotide. Example EMBL: mam*. Example BOLD: Actinopterygii
-q QUERY, --query QUERY
NCBI query search to limit portion of database to be downloaded. Example: "16S[All Fields] AND ("1"[SLEN] : "50000"[SLEN])"
-p SPECIES, --species SPECIES
species to be downloaded, either as a string separated by "+" or as a list in a .txt file
-m MARKER, --marker MARKER
genetic marker to download for BOLD
-o OUTPUT, --output OUTPUT
output file name
-k ORIG, --keep_original ORIG
keep original downloaded file, default = "no"
-d DISCARD, --discard DISCARD
output filename for sequences with incorrect formatting, default = not saved
-g BOLDGAP, --boldgap BOLDGAP
incorporate or discard sequences with gaps from BOLD (DISCARD/INCORPORATE). Default = DISCARD
-e EMAIL, --email EMAIL
email address to connect to NCBI servers
-b BATCHSIZE, --batchsize BATCHSIZE
number of sequences downloaded from NCBI per iteration. Default = 5000
2.3 Step 1: download sequence data#
As a first step, we will download the 16S shark sequences from the NCBI online repository, as well as all sequences from the Japanese MitoFish database. Downloading the Japanese MitoFish databases is quite straightforward, as it is a single file. We can use the crabs db_download -s mitofish command to accomplish this. The only other parameter we need to provide is the output file name.
crabs db_download -s mitofish -o sequenceData/7-refdb/mitofish.fasta
Output
downloading sequences from the MitoFish database
complete_partial_mitogenomes.zip 100%[===============================================================================>] 79.27M 6.05MB/s in 13s
formatting mito-all to CRABS format
93%|██████████████████████████████████████████████████████████████████████████████████████████████▊ | 781202771/840651415 [00:05<00:00, 140055187.99it/s]
found 0 sequences with incorrect accession format
written 825365 sequences to sequenceData/7-refdb/mitofish.fasta
To download data from the NCBI servers, we need to provide a few additional parameters to CRABS. Maybe one of the trickiest ones to understand is the --query parameter. We can get the input string for this parameter from the NCBI website, as shown on the screenshot below.
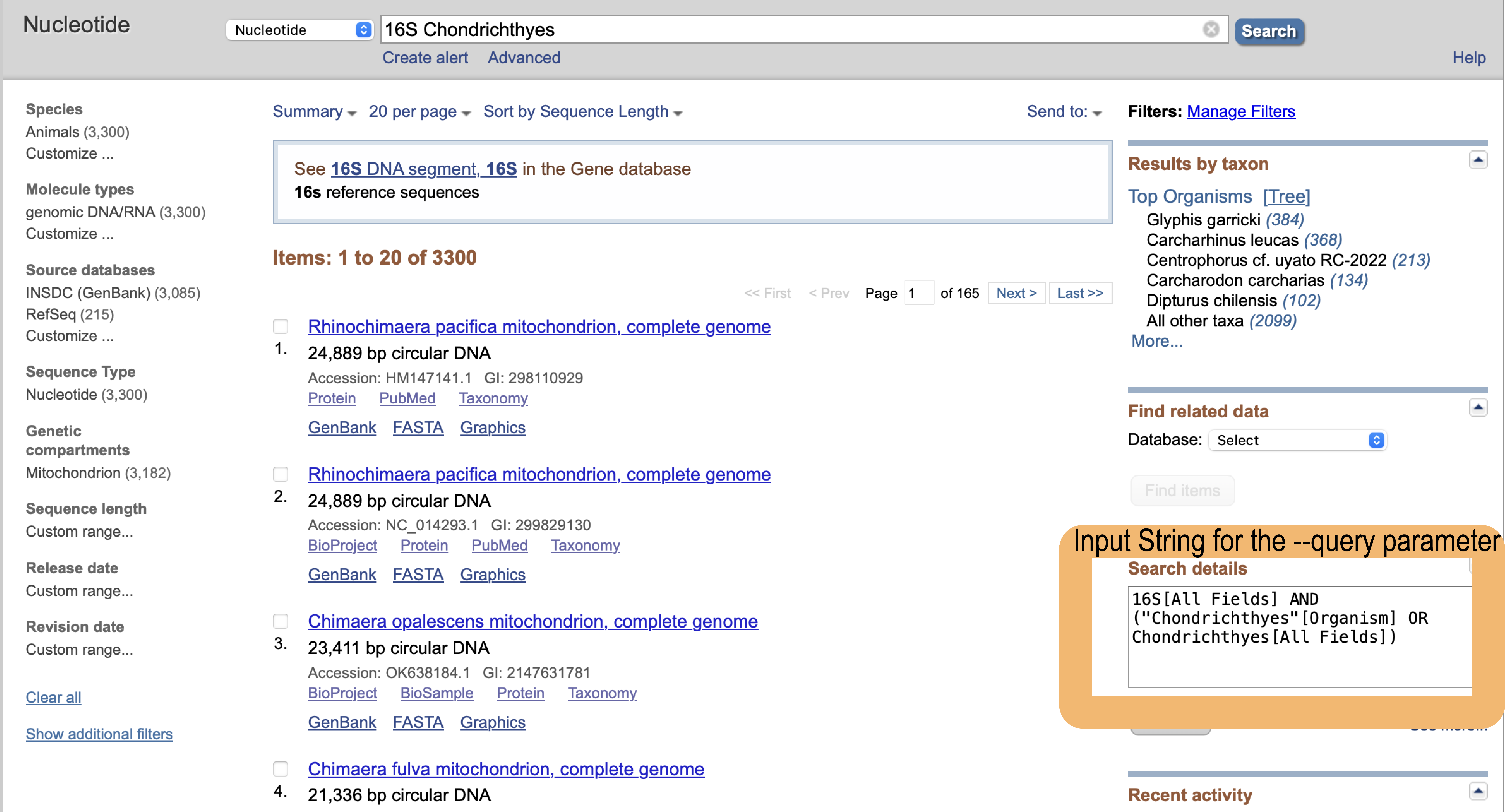
Fig. 22 : A screenshot on how to obtain the query string to download sequence data from the NCBI servers through CRABS.#
crabs db_download -s ncbi -db nucleotide -q '16S[All Fields] AND ("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields])' -o sequenceData/7-refdb/ncbi16Schondrichthyes.fasta -e gjeunen@gmail.com
Output
downloading sequences from NCBI
"--species" parameter not provided, downloading NCBI data based solely on "--query": 16S[All Fields] AND ("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields])
looking up the number of sequences that match the query
found 3300 number of sequences matching the query
starting the download
temp_ncbi16Schondrichthyes.fasta_1.fasta [ <=> ] 28.92M 310KB/s in 33s
formatting the downloaded sequencing file to CRABS format
97%|████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████▋ | 29553742/30322979 [00:00<00:00, 275397805.14it/s]
found 0 sequences with incorrect accession format
written 3300 sequences to ncbi16Schondrichthyes.fasta
When downloading data from online repositories, CRABS will immediately reformat the data to a simple 2-line fasta format whereby the header information only contains the unique identifier (accession number or a CRABS number if the accession number is not available). If you have generated your own reference barcodes, you can import them into CRABS format using the db_import command.
head -n 2 sequenceData/7-refdb/ncbi16Schondrichthyes.fasta
Output
>OR575552
GCTAGCGTAGCTTATCGAAAGCATGGCACTGAAGATGCTAAGATGAAAAATAAAAATTTCCGCGGGCATGAGAGGCTTGGTCCTGGCCTTAGTGTTAATTGTAATTAGGATTATACATGCAAGTCTCAGCACACCTGTGAGAATGCCCTCATTAGTCTATTAACTAATCAGGAGCTGGTATCAGGCACACGCACGTAGCCCAAGACACCTTGCTAAGCCACACCCCCAAGGGATTTCAGCAGTAATAAACATTGATTTATAAGCGCAAGCTTGAATCAGTTAGGATTAACTGGGTTGGTAAATCTCGTGCCAGCCACCGCGGTTATACGAGTAACCCTTATTAACACTCTACCGGCGTAAAGAGTGGTTTAAGAATTTTTCCCTATGAACTAAAGTTAAGACCCCATTAAGCTGTTATACGCCCTCATGGGTGGAATCACCAACAACGAAAGTAACTTTACAGCACCAGAAAACCTTGACCCCACGACAGTTGAGCCCCAAACTAGGATTAGATACCCTACTATGCTTAACCCCAAACTTAGATAATAACCCACCTATTATCCGCCAGAGTACTACAAGCGCTAGCTTAAAACCCAAAGGACTTGGCGGTGCCTCAGACCCACCTAGAGGAGCCTGTTCTATAATCGATACTCCCCGTTTAACCTCACCACATCTTGCCAATACCGCCTATATACCGCCGTCGTCAGCCCACCCTGTGAGGGTTAAGAAGTAAGCAAAAAGAATTAACTTCCACACGTCAGGTCAAGGTGTAGCGAATGAAGTGGAAAGAAATGGGCTACATTTTCTATTAAGAAAACACGAACAGTAAACTGAAAAATTACTCGAAGGTGGATTTAGCAGTAAGAAAAATTAGAGTATTTTTCTGAAGCCGGCTCTGAAGCGCGCACACACCGCCCGTCACTCTCCTTAAAAAAATACTAATTTATACTTAAACAAAATTATTTTACAAGAGGAGGCAAGTCGTAACATGGTAAGTGTACTGGAAAGTGCACTTGGAATCAAAATGTAGCTAATTTAGTAAAGCACCTCCCTTACACCGAGGAAAAACCCGTGCAATTCGAGTCATTCTGAACATTAAAGCTAGCCTAATTATCATATAAACACAACCCCATTAATTACACACCATTTTTACCAGAACTAAAACATTTTACACTTTTTAGTATAGGCGATAGAACGATAAACCAGCGCAATAGATTAAGTACCGCAAGGGAAAACTGAAAAAGAAATGAAACAAATCATTAAAGTAATAAAAAGCAGAGATTTAACCTCGTACCTTTTGCATCATGATTTAGCTAGATCAATTAGGCAAAAAGATCTTAAGTCTACCTTCCCGAAACTAAACGAGCTACTCCGAAGCAGCAAATTAGAGCCAACCCGTCTCTGTGGCAAAAGAGTGGGAAGACTTCCGAGTAGTGGTGACAGACCTATCGAGTTTAGTGATAGCTGGTTACCCAAAAAAAGAACTTAAATTCTGCATTAATTTTTTACCCACCAATAAGAACATCTCCACAAGATGGTAAATAAGAATTAATAGTTATTTAGAAGAGGTACAGCCCTTCTAAACCAAGATACAACTTTTAAAGGCGGGTAATGATCACATTTTACTAAGGTTTACCCACCAGTAGGCCTGAAAGCAGCCATCTGTAAAGTAAGCGTCACAGCTCCAGCTCGACATAAACCTATAATCTAGATACCACCTCACAACCCCCTACAAAATACTGGGTTATTTTATACAACTATAAAAGAACTTATGCTAAAATGAGTAATAAGAGACCAAACCTCTCCCAACACAAGTGTACATCAGAAAGAATTAAATCACTGATAATTAAACGAATCCAAACTGAGGGTATAATATTAACTCACCACCAACTAGAAAACCTTATTTAACCATTCGTTAATCCTACACAGGAGTGTCCTCAAGGAAAGATTAAAAGAAAATAAAGGAACTCGGCAAACACAAACTCCGCCTGTTTACCAAAAACATCGCCTCTTGCTTACTATAAGAGGTCCCGCCTGCCCTGTGACAATGTTCAACGGCCGCGGTATTTTGACCGTGCGAAGGTAGCGTAATCACTTGTCTTTTAAATGAAGACTTGTATGAAAGGCATCACGAGAGTTTAACTGTCTCTATTTTCTAATCAATGAAATTGATCTTCTCGTGCAGAAGCGAGAATAACACCATTAGACGAGAAGACCCTATGGAGCTTCAAACACTTAAATTAATTATGTAAAATTTTTAACTCTCCGGAGAAAAACCACCCATATAATACCCCTAATTTAACTGTTTTTGGTTGGGGTGACCAGGGGGAAAAAATTATCCCCCCCATCGATTGAGTACTCAGTACTTAAAAATTAGAATGACAACTCTAATTAATAAAACATTTACCGAAACATGACCCAGAATTTATTCTGATCAATGAACCAAGTTACCCTAGGGATAACAGCGCAATCCTTTCCCAGAGTCCCTATCGACGAAAGGGTTTACGACCTCGATGTTGGATCAGGACATCCTAATGGTGCAACCGCTATTAAGGGCTCGTTTGTTCAACGATTAATAGTCCTACGTGATCTGAGTTCAGACCGGAGAAATCCAGGTCAGTTTCTATCTATGAATTAATTTTTCCTAGTACGAAAGGACCGGAAAGATGGAGCCAATACCATGAGCACGCTCCATTTTCACCTACTGAAATAAACTAAATTAGGTAAGAAAACATTACCTAAAACCCGAGACAAGGGTTATTGGGGTGGCAGAGCCTGGTAAATGCAAAAGACCTAAATCCTTTAATCCAGAGGTTCAAATCCTCTCCCCAATTATGCTCCAATACATTTTACTGTACCTCGTAAATCCTCTTGCCTACATCATCCCAGTTCTTTTAGCAACAGCCTTCCTAACCTTGGTCGAACGAAAAATCCTAGGCTACATACAATTCCGCAAGGGCCCCAACATCGTAGGACCATACGGCCTCCTTCAACCAATTGCAGACGGCGTAAAACTTTTTATTAAAGAACCAGTCCGCCCATCAACATCCTCACCCCTCTTGTTCCTAGCAACCCCAACCCTAGCACTAACCCTAGCCCTCCTTTTATGAATACCTCTCCCCCTACCACACTCAATCATCAACTTAAACTTAGGTCTACTATTCATCCTAGCCATCTCAAGCCTAACTGTTTACACAATCCTAGGCTCAGGATGAGCATCCAACTCAAAATATGCCCTCATAGGAGCACTACGAGCCGTAGCACAAACTATTTCCTATGAAGTAACTCTCGGCCTCATCCTCCTATCGATAATCATCTTCACGGGTGGATTCACACTCCACACATTCAACCTAACTCAGGAGGCAATTTGATTGTTTATCCCAGGCTGACCACTTGCTATAATATGGTATATCTCAACCCTGGCAGAAACCAACCGAGCACCGTTCGACTTAACCGAGGGAGAATCAGAACTGGTCTCAGGGTTTAATGTTGAATATGCAGCTGGCTCATTTGCCTTATTCTTCTTAGCCGAATACACAAACATCCTAATAATAAATACACTCTCAGTGATTCTATTTTTAGGCTCCTCCTATAACCCCTCTATACCACAAATCTCCACATTTTACTTAATGATAAAAGCAACACTACTCACCCTAATTTTTTTATGGGTGCGAGCATCATACCCCCGCTTCCGCTATGACCAACTTATACACCTAGTGTGAAAAAATTTCCTGCCACTCACCCTAGCCCTCATCCTATGACACGCAACCCTACCGCTCGCTGTAGTGAGCCTCCCCCCCGCAACCTAAACGGAAACGTGCCTGAATTAAAGGACCACTTTGATAGAGTGGGTCATGAGAGTTAAAATCCCTCCGCTTCCTCCTAGAAAAACAGGACTTGAACCTGCACCCGAGAGATCAAAACTCCCGGTACTTCCTATTATACTATCTCCTAAGTAAAGTCAGCTAATTAAGCTTTTGGGCCCATACCCCAACGATGTTGGTTAAAATCCTTCCTCTACTAATGAACCCTCTAGTACTAACCACTATTATCTCAAGCCTTGGCCTAGGAACCACAATAACATTCATCGGATCCCATTGACTTCTAGTTTGAATAGGTCTTGAAATTAACACCTTAGCCATTATTCCCCTAATAATCTACCACCACCACCCGCGAGCAGTAGAAGCAACCACAAAATATTTCCTAACACAAGCAACTGCCTCCGCCCTACTTTTATTTGCTAGCATCACAAACGCCTGAGACTTGGGCGAATGAAATTTAATTGAATTAATTAACCCAACCTCTGCCACACTAGCAACCATCGCATTAGCTTTAAAAATCGGACTTGCACCACTACACTTTTGACTACCCGAGGTCCTTCAGGGACTAAACCTAACCACAGGCCTGGTCCTAGCCACGTGACAAAAACTCGCCCCATTTGCCATCCTCCTACAACTCCACCCCATCCTTGACTCAAACTTACTGTTATTCCTAGGCATTTTATCAATCATTGTCGGAGGTTGGGGGGGACTCAACCAAACCCAACTACGAAAAATTCTGGCTTACTCATCAATCGCCCACTTGGGATGAATAATTACAATCCTACACTACTCCCCAAACCTCACCAAGCTCAACCTCACTTTATATATTATTATGACCCTCACAACCTTCCTCCTATTTAAACTATTCGACTCCACCAAAATCAACTCCATCGCATCCTCCTCAATCAAATCCCCCCTCCTTTCTGTTATTACATTAATAACCCTCCTCTCCCTAGCCGGCCTGCCACCCCTCTCCGGATTTATACCAAAATGATTAATTTTACAAGAACTAGTTAAACAAAACCTAGCCGCCCCTGCCACAATCATAGCCCTCGCAACACTACTAAGCTTGTTCTTCTACCTACGCCTATCTTACTCAACAACCCTAACAATATCCCCAAATTCTATCTACTTATCATCATCTTGACGAACAAAATCAATACAACAAAATCTTCCCCTAATAATAACCGCCTCTATTACCATCCTACTCCTGCCACTAACCCCAGCTATCTTTATATTAACCCTCTAAGAAATTTAGGCTAAACAGACCAAAAGCCTTCAAAGCTTTCAGTAGGAGTGGAAATCCCCTAATTTCTGCTAAGATTTGCAAGACTTTATCTCACATCTTCTGAATGCAACCCAGATACTTTCATTAAGCTAAAACCTTCTAGATAAATAGGCCTTGATCCTACAAAATCTCAGTTAACAGCTAAGCGTTCAATCCAGCGAACTTTTATCTAGGCTTCCTCCCGCCGTCAATACGGGGAGGCGGGATGAAGCCCCGGGAGGAGGAAACCTCCGGTTTTGGATTTGCAATCCAACGTAACGTCTACTGCAGGACTTGGTAAGAAGAGGAATTTAACCCCTGTTTACGGGGTTACAACCCGCCACTTAGTTCTCAGCCATCTTACCTGTGGCAATTAACCGTTGATTCTTTTCTACAAATCATAAAGATATCGGCACCCTATACTTGATCTTTGGTGCATGGGCAGGAATAGTAGGCACAGCCCTTAGCCTACTAATTCGAGCTGAATTAAGTCAGCCAGGATCCCTTCTTGGTGATGATCAAATCTACAATGTAATTGTGACTGCTCACGCTTTCGTAATAATCTTTTTTATAGTTATACCTGTAATAATTGGTGGGTTTGGAAATTGACTAGTGCCTTTAATAATTGGCGCACCAGATATAGCCTTCCCACGAATAAATAACATAAGCTTTTGATTACTACCCCCGTCGCTCCTTCTACTCTTAGCCTCCGCCGGCGTAGAAGCAGGAGCTGGAACTGGTTGAACGGTCTACCCACCACTCGCCAGTAATATAGCCCACGCGGGGGCATCTGTAGACTTAGCAATTTTTTCACTCCATCTAGCTGGTATCTCCTCAATTCTAGCCTCTATTAACTTTATCACAACCATCATCAACATGAAACCACCCGCCATTTCTCAATACCAAACACCACTTTTTGTTTGATCCATCTTAGTGACAACTGTCCTGCTTCTCCTGGCCCTCCCAGTCCTTGCAGCTGCAATTACAATACTGCTAACTGACCGTAATTTAAATACAACATTTTTTGACCCGTCAGGGGGAGGAGACCCCATCCTATATCAACACTTATTTTGATTCTTTGGTCACCCAGAAGTTTACATTTTAATTTTACCAGGCTTCGGAATAATTTCACACGTGGTAGCCTACTACTCAGGGAAAAAAGAACCTTTTGGCTACATGGGAATAGTCTGAGCAATAATAGCAATCGGTTTACTAGGCTTTATTGTCTGAGCCCATCATATATTTACGGTTGGGATAGACGTCGACACCCGCGCCTACTTCACATCAGCAACAATAATTATTGCTATCCCAACAGGCGTGAAAGTTTTTAGTTGACTAGCGACCCTGCATGGGGGGTCAATTAAATGAGAAACCCCACTCCTTTGGGCTTTAGGTTTCATTTTCTTATTCACAGTTGGCGGCCTCACTGGAATTGTTTTAGCCAACTCCTCTTTAGACATCGTTCTCCATGACACTTATTATGTAGTAGCCCACTTCCACTATGTTTTATCAATAGGGGCAGTATTTGCAATTATAGCGGGCTTCATCCACTGATTCCCTCTATTCTCGGGGTACACCCTTCACTCAACATGAACAAAAACCCAATTCCTAGTAATATTTATTGGGGTAAATTTAACTTTCTTCCCCCAACACTTCTTAGGCTTAGCCGGCATACCTCGACGGTACTCAGACTACCCAGATGCATACGCCCTTTGAAACACGGTTTCATCAATTGGCTCATTAATTTCATTAGTTGCCGTAATTATGTTCCTATTTATTATTTGAGAAGCATTTGCCGCTAAACGCGAAGTTCTATCTGTAGAATTACCGCACACAAACGTGGAGTGACTACACGGCTGCCCGCCACCCTATCACACCTACGAAGAGCCAGCATTTGTACAAATCCAACGAACCCATTTCTAACAAGGAAGGGAGGAATTGAACCCCCGTATATTGATTTCAAGTCAATCACATCACCACTCTGTCACTTTCTTTAAGATTCTAGTAAAATATATTACACTGCCTTGTCAGGGCATAGTCATGAGTTTAAACCTCGTGTATCTTATTTTACAATGGCACACCCAGCACAATTAGGATTTCAAGACGCAGCCTCCCCAGTTATGGAAGAACTTCTACATTTTCATGACCACACACTAATAATCGTATTTTTAATTAGTACTTTGGTCCTTTATATTATCTTAGCAATGGTATCCACTAAACTCACAAACAAATACATTTTAGATTCACAAGAAATCGAAATAGTGTGAACTATTCTCCCTGCTGTGATTCTTATTATAATCGCCCTTCCATCATTACGAATTTTATACCTCATAGATGAGATCAACGACCCACACCTAACCATTAAAGCCATGGGCCACCAATGATATTGAAGCTATGAATACACAGACTACGAAGACCTGGGGTTCGACTCCTACATAATCCAAACTCAAGATTTAGCCCCCGGCCAATTTCGCCTGCTAGAAACTGACCATCGGATAGTTGTACCAATAGAATCCCCAATTCGTGTCTTAGTATCCGCAGAAGATGTACTTCACTCATGAACAGTTCCAGCCCTGGGAGTCAAAATAGATGCAGTACCGGGACGACTAAATCAAACAGCCTTTATTGTCTCCCGCCCAGGTGTGTACTACGGTCAATGCTCGGAAATTTGCGGCGCCAATCATAGCTTTATACCCATCGTAGTTGAAGCAGTCCCACTAGAACACTTTGAATCCTGATCTTCATTAATACTAGAAGAAGCCTCACTAAGAAGCTATACTGGGTTTTAGCATTAGCCTTTTAAGCTAAAAATTGGTGACTCCCCACCACCCTTAGTGACATGCCCCAGTTAAACCCAAATCCATGATTTGCCATTTTCATACTCTCATGAATTTTCTTCCTAGCTATTTTACCAAAAAAAGTGATAACCTATCTATTTAATAATAACCCAACACCAAAAAGCGCTGAAAAACCTAAACCTGAACCCTGAAACTGACCATGAACCTAAGCTTTTTTGACCAATTCTTGAGCCCATCACTTCTTGGAGTCCCACTGATTGCCCTAGCAATCATAATCCCATGGTTAATCTTCCCAGCCCCAACAAACCGCTGACTTAATAACCGCCTCGTGACAATCCAATCATGATTCATCAACCGCTTTACACATCAGCTAATACAACCTATAAATTTTGGAGGTCACAAGTGAGCCACCATTTTAACAGCCCTCATACTATTCTTGATTACTGTTAACCTTCTTGGCCTACTCCCATACACCTTCACACCCACAACCCAATTATCACTTAACATAGCATTTGCCATCCCACTTTGATTAACAACCGTCCTCATCGGAATACTTAACCAACCAACAATTGCCCTTGGACACCTTCTCCCAGAAGGCACTCCGACCCCACTAATTCCAATCCTAATTATCATCGAAACAATTAGCTTGTTCATCCGACCCCTAGCACTAGGCGTCCGACTTACTGCAAACCTGACAGCCGGCCACCTCCTCATACAACTGATTGCCACCGCAGCCTTCGTACTAATTACCATCATGCCAGCCGTAGCCATACTAACCTCATTAATCTTATTCCTATTAACAATTTTAGAAGTAGCCGTGGCAATAATTCAAGCTTACGTATTTGTCCTATTACTAAGTCTCTATCTACAAGAAAACGTCTAATGGCTCACCAAGCACATGCATATCATATAGTCGACCCCAGCCCATGACCCCTAACAGGAGCTGTTGCTGCCTTACTAATAACATCAGGCCTAGCCGTCTGATTTCACTTCCACTCTTTATACCTCCTCTACCTCGGATTAATTCTTCTATTCTTGACCATGATTCAATGGTGGCGAGATGTCATTCGAGAAGGAACATTTCAAGGCCACCACACACCACCTGTTCAGAAAGGACTTCGCTATGGAATAATTTTATTTATTACATCAGAAGTTTTCTTTTTCCTTGGCTTCTTCTGAGCCTTCTACCACTCAAGCCTTGCCCCCACACCAGAACTAGGTGGATGCTGACCACCCACAGGAATCTACCCACTAGACCCCTTCGAAGTCCCCTTATTAAACACTGCAGTCCTTCTTGCATCCGGGGTCACAGTAACCTGAGCCCACCACAGCTTAATAGAAGGTAACCGAAAAGAAGCAATCCAAGCCCTAACTCTAACCGTATTATTAGGATTTTATTTTACAGCTCTTCAAGCCATAGAATACTACGAAGCTCCATTCACCATTGCTGATGGGGTTTATGGTTCAACATTCTTTGTTGCCACAGGTTTTCATGGTCTCCATGTTATTATTGGTTCAACATTTTTAATAGTTTGTCTAATACGACAAATCCAATATCATTTTACATCAGAACATCACTTTGGCTTTGAAGCCGCAGCATGATACTGACATTTTGTCGATGTAGTATGATTATTCCTTTATGTTTCCATCTACTGATGAGGTTCATAATTACTTTTCTAGTAAAAACTAGTACAAGTGATTTCCAATTACTTAATCTTGGTTAAAATCCAAGGGAAAGTAATGAACCTCATCATGTCATCAGTCGCAACCACGGCCCTCATTTCCCTAATCCTCGCTTTCATAGCATTTTGACTCCCATCACTAAACCCAGACAACGAAAAACTATCACCCTACGAATGCGGGTTTGACCCACTAGGGAGTGCACGCCTCCCGTTTTCATTACGTTTTTTCCTCGTGGCAATTTTATTTCTCCTGTTCGACCTCGAAATCGCCCTCCTCCTCCCACTGCCCTGAGGGAATCAACTATTAACACCACCCACTTCACTTCTATGAGCAACAAGCATCATTATTCTATTAACCCTGGGCCTCGTCTACGAGTGGTTTCAAGGTGGCCTTGAATGAGCAGAGTGGGTATTTAGTCCAAACAAGACCACTAATTTCGGCTTAGTAAATTATGGCGAAACACCATAAATACCTTATGTCCCCCATCTACTTCAGCTTTAGCTCAGCATTCACCATAGGTTTAATAGGCCTAGCATTCAACCGATCACACCTCTTATCAGCCTTACTTTGTTTAGAGGGAATAATATTATCCTTGTTTATTGCTATTGCCCTCTGATCAATAACACTAAACTCAGCATCATGCTCAATTACCCCGATAATCATCCTCACTTTCTCTGCCTGTGAAGCAAGCGCAGGTTTAGCCATATTGGTGGCAACCACACGAACCCACGGTTCCGACCACTTACAAAACCTGAGTCTTCTACAATGCTAAAAATTTTAATTCCAACAATCATACTATTCCCCACCACATGACTACTACCCATAAAATGACTGTGACCCGTTATAACCTCCCACACCCTTCTAATCGCACTACTTAGCCTACTATGATTTAAATGAAGCACGGACATTGGGTGAGACTTCTCCAATCACTACATAGGTGTAGACCCACTCTCAGCCCCCCTACTTGTCCTCACCTGTTGGCTCCTCCCATTAATAATCCTGGCCAGTCAAAACCACATATCCATAGAGCCGATTATTCGCCAACGAACTTACATCACACTTCTAATTTCACTACAAACCTTCCTCATCCTAGCATTCAGTGCAACAGAATTAATCATATTTTACATCATATTTGAAGCCACACTTATCCCCACCCTTATCATCATCACTCGATGGGGAAACCAAACCGAACGATTAAATGCAGGAATTTACTTTCTATTCTACACCCTAATTGGCTCCCTTCCTTTACTAGTTGCCCTTCTACTCATACAAAACAACTTGGGCTCATTGTCAATAATTATTATTCAACACCCACAACCACTCAACCTCAACACATGAGCCAACAAACTCTGATGGGCCGCCTGCCTTATCGCCTTCTTAGTAAAAATACCCCTCTATGGTGTTCACCTTTGACTCCCAAAAGCCCATGTTGAAGCCCCCATTGCAGGGTCAATAATCTTAGCCGCCGTACTACTAAAACTTGGTGGTTACGGAATAATACGAATTATTGTTATACTTGACCCCACCACCAAAGAAATAGCATACCCCTTTTTAATCTTAGCCGTCTGAGGGGTAATTATAACAAGCTCAATTTGCCTTCGACAAACTGACCTAAAATCCCTAATCGCTTACTCATCAGTAAGCCACATAGGACTAGTCGCAGCAGCAATTATAATCCAAACACCATGAAGCTTCGCAGGAGCAATTACACTAATAATTGCACATGGGTTAGTCTCGTCCGCTCTCTTCTGCCTAGCAAATACCAATTATGAACGCATCCACAGCCGAACCCTACTCTTAGCACGTGGCATTCAAATTATCCTCCCACTAATGGCAACCTGGTGATTCATTACTAACCTTGCCAACTTAGCTCTACCACCAACCCCTAATTTAATAGGAGAACTAATCATCATCTCCTCATTATTCAATTGATCCAACTGAACAATTTTACTAACGGGACTTGGAGTACTAATTACAGCATTATACTCCCTATACATGTTCTTAATAACCCAACGAGGCCCCACACCTAAACACCTGCTATCCTTAAACCCATCCCACACACGAGAGCACCTCCTCCTCAACCTTCACCTCACCCCCATATTACTATTAATCCTCAAACCCGACCTTATCTGAGGATGAACCTTTTGCCACTATAGTTTAACCAAAACATTAGATTGTGGTTCTAAAAACAAAAGTTAAAACCTTTTTAGTAGCCGAGAGTGGTCTGGGACACAAAGAACTGCTAATTCTTTCTACCACGGTCCGAATCCGTGGCTCACTCAGCCCTTGAAAGATAACAGCCATCTATTGGTCTTAGGAACCAAAAACTCTTAGTGCAACTCTAAGCAAGAGCTATGAGCACAACTATTTTTAACTCAACCTTTCTTCTTATCTTCATCATCCTACTGTACCCACTCATTTCATCACTCTCACCTAAAGAGCTCAACCCAAACTGATCATCTTCACATGTAAAAACCGCTGTAAAAATCTCTTTCTTCATCAGCCTTATCCCACTACTCATTCATCTAGATCAAGGCCTAGAATCAACCACAACCAACTGAAACTGAATCAATATTGGACCTTTTGATATCAACATAAGTTTTATATTTGACTCATACTCAATTATCTTCACCCCAGTAGCCCTTTACGTTACTTGATCAATCCTTGAATTTGCCATCTGATACATACACTCAGATCCAAACATTAACCGCTTCTTCAAATATCTTCTCCTGTTTTTAATTTCCATGATTATCTTAGTAACAGCCAATAATATATTTCAACTATTTATTGGCTGAGAGGGGGTCGGAATCATATCTTTCCTTTTAATTGGTTGGTGGTTCAGTCGAGCAGACGCAAATACTGCGGCCCTACAGGCCGTGATTTACAACCGTGTGGGGGATATTGGACTCATCTTAAGCATAGCCTGATTAGCCACAAACCTAAACTCATGAGAAATTCAACAGCTCTTTATCCTATCAAAAGATAAAGACCTCACCCTGCCTCTTATTGGCTTAGTACTAGCCGCAGCGGGAAAATCTGCACAGTTTGGACTTCACCCGTGACTACCTTCCGCCATAGAAGGGCCCACACCAGTCTCTGCCCTACTCCACTCCAGCACAATAGTGGTAGCCGGCATTTTTCTACTAATTCGCCTTCACCCATTAATCCAGGACAACCAACTAATCTTAACAACATGTTTATGTTTGGGGGCCCTAACCACCCTATTTACTGCCGCATGCGCACTAACACAAAACGACATTAAGAAAATTGTTGCCTTCTCAACATCAAGCCAACTAGGCCTAATAATAGTCACAATCGGACTAAACCAACCACAGTTGGCGTTTCTTCATATCTGCACACATGCTTTCTTTAAAGCTATACTTTTCCTCTGTTCCGGTTCGATTATCCACAGCTTAAATGATGAACAAGACATCCGAAAAATAGGCGGCCTTCATAAACTTCTACCGTTCACCTCTTCATCACTAACTGTGGGCAGCCTTGCCCTCACAGGGATACCATTCCTGTCAGGATTTTTTTCAAAAGATGCCATTATTGAATCCATAAACACCTCCCATTTAAACGCCTGGGCCCTAATTTTAACTCTTATTGCAACATCATTCACCGCCATTTATAGCCTCCGCCTTATCTTTTTCGCCCTAATAAAATTTCCGCGATTCTCCACCCTATCCCCAATCAATGAAAATAACACCTTAGTTATAAACCCCGTCAAACGCCTCGCATACGGAAGCATCTTTGCTGGGTTAATCATTACCTTTAACCTACCGCCAGCAAAAACTCAAATCATAACTATAACCCCACTATTAAAACTTTCTGCCCTCCTGGTAACAATTATCGGTTTACTTCTAGCACTAGAACTAACCAACTTAACTAACACCCAACTAAAAATTACCCCCACCACATCTACCCACCACTTCTCCAACATGCTTGGATATTTTCCAATAGTTGTTCACCGACTCACACCAAAAATTGACCTAAAATGAGCTCAACACATCTCAACACACCTTATTGACCTCACTTGAACTGAAAAAATTGGACCAAAAAGCACCATTATTCAACAAACACCATTAATTAAATTATCAACCCAACCACAACAAGGAATAATCAAAACCTACCTGATACTGCTCTTTTTAACACTAACACTAGCCACCCTAATCACCACCGCTTAAACAACACGCAAACTCCCATATGATAAACCACGAGTTAATTCTAAAACCACAAACAACGTTAAAAGCAGAACTCACCCGCCCAACACTAATATCCAACCACCGTGGGAGTATAACAAAGCTACGCCACCCAGATCTCCTCGAATTATCTCCAGAGTACTTATTTCCTCAACCCCAGCCCAACTTAATCCCAACCATTTATTAAAAAAATATTGACCTACAAAAAAAAGGCCTAATAAATACACACCAACATACAATAACACAGACCAATCGGCCCACGTCTCTGGGTATGGTTCGGCAGCAAGTGCTGCTGTATAAGCAAACACGACCAATATCCCCCCCAAATAAATTAGAAATAAGACTAAAGATAAAAAAGAACCCCCATGTCCCACTAATAAACCACACCCCACTCCAGCAACCACCACCAAACCCAAAGCCGCAAAATATGGGGAGGGGTTTGACGCTACTGCTATCAATCCTAAAATTAAACCCACTAATATTACAAACATAAAATAAATCATTATTCTCACCTGGGCTCTAACCAAGACCGACAACTTGAAAAACTATCGTTGTTCATTCAACTACAAGAATAATAATGACCACCAATATCCGAAAAACCCACCCACTCATTAAAATCGCCAACCACGCTTTAGTCGATTTACCCACACCATCCAACATCTCGATCTGATGGAATTTTGGTTCGCTACTTGGACTTTGCTTATTTATCCAAATTCTCACTGGATTATTTTTAGCCATACACTACACCGCAGACATTTCCACAGCCTTCTCATCAGTGATTCACATCTGTCGTGATGTAAACTATGGCTGACTAATTCGTAACATTCACTCTAACGGAGCCTCAATATTCTTCATTTGTGTTTACTTACACATTGCCCGAGGACTATACTACGGCTCCTACCTCTTTAAAGAAGTGTGAAACATCGGAGTTATTCTACTGTTCCTACTTATAGCAACAGCCTTTGTGGGCTACGTCCTACCATGAGGGCAAATATCCTTCTGAGGTGCCACAGTCATCACCAACCTCCTGTCCGCCCTCCCTTATATCGGGGACACATTAGTACAGTGGGTTTGGGGGGGTTTTTCAATTGATAACGCCACCCTAACACGCTTCTTCGCATTTCACTTCCTCCTACCATTTTTAATCCTAGGTTTAACCCTAATTCATCTACTATTCCTACACGAAACTGGCTCCAACAACCCCCTAGGCCTAAACTCTGACATAGATAAAATTCCATTCCACCCCTACTTCACATACAAAGATCTCTTGGGTTTCCTTATAATAACCATTTTATTAACCACATTGGCTTTATTCTCACCCAACCTTTTAGGTGACGCCGAAAATTTCATTCCCGCAAACCCACTCGTCACACCACCACACATCAAACCAGAGTGGTATTTCCTATTCGCCTACGCCATTCTTCGATCAATTCCTAACAAACTAGGTGGGGTATTAGCCTTAGCATTCTCAATTTTCATCCTAATACTAGTCCCCATACTTCACACCTCAAAACAACGAAGTAGCAGCTTCCGGCCCCTTACACAAATTCTCCTTTGAACACTCGTAGCTAACGCGATCATTCTAACATGAATTGGAGGACAACCCGTTGAACAACCATTTATCCTTGTTGGCCAAATTGCTTCCACTACTTATTTTTCCTTGTTTCTCATTATCATTCCGGCAACTAGCTGGTATGAAAACAAAATGCTCAACTTAAATTAATGTTTTGGTGGCTAAAATAAAAAGCGTCGGCCTTGTAAGTCGAAGACCGGAGGTGCAAATCCTCCCCACAACACATCAGGGAAAGGAGGGTTAAACTCCTGCCATTGGCTCCCAAAGCCAAGATTCTGCCTAAACTGCCCCCTGATCGCTAAGATCCCGCGAAAGCGGGGTTTTTTTGGAGTAAGTCAGTTGTACATATAAGTGATATAGCCCCCATACCCTAATATACCACATACGACATACATGTATAATCACCATATATAGACTTTCCCCATCTATATTACATCTATATGTTTAATACTCATTCATCTATAATCCTCACCCTCATTACATATATGATTAACCCACATTTCACTAAGATCAATAAATTCATTACATTAAATTTTCATTAACCCACATTTCCTTAAGATCACCGGATCCATTACATAAAATCCTTAACCCTCATAAACATAAGATCAATTATTTAACTAGGGAATATAAGAACCGTACAACGATCTCTTAGGTAAATATAGTGTACGGTTTGTGGTACGGTTCTCGAGATATCCCCTATAGGTGATCAAATGCTGGCATTTGGCACCCTTAAAGGGCTGACAGTTCAGACGTATCATGATCTCCAGACCGCTAGTTCCCTTAGTTGTCATTCAACTCTTAACCGTCTCAAGATTTACTGTCCGCCCTGTTTTTTTTTGGGGGGATGCGAGCAATCGCTAAGACCCGGAAGGCCCCAGGCATGGTCATACAGGTAAACCCGAACTCATCTAGACCTTACTCTCTTATTTAACAATACTGAATTCTTGTAATAAGATTGTCAAGTTGACCATATAGACAAAGGCATGGAGATCCTAATATTATAAGTGACACAATTTGGTTTCGTGCGTAATTAAACTTTCGTTCAATATGCACATATCATTTATCCTCATTTCTGTTTGGTAATCGATATCTATTAATGAGAATTAACATTCAACCTATTTGGTACTAACGTCCCTATTCGAGCATAGTTGATAGTAATTGCGCAATATTTCTGTAATATTACCCCGGGTGACGTAACGGAAGACGACGAATTATTAACTTTGGTTTTCTGGGAAAAACCCCCCCTCCCCCCTAATATACAAACTGACTTTTCTCGAAAAACCCCAAAAACGAGGGCCGAATGCATATTCCCATTTTTTAGCGTGTGAAAATATTTGCTATACATTGTTGCAACATGTTTTGCACAATGTGAC
2.4 Step 2: merge downloaded sequences#
Once we have downloaded the sequences from the MitoFish and NCBI repositories, we can combine both files using the db_merge command. Since there will be overlap in the data we downloaded between the online repositories, we can specify the --uniq parameter to only keep unique accession numbers.
crabs db_merge -o sequenceData/7-refdb/mergedDB.fasta -u yes -i sequenceData/7-refdb/ncbi16Schondrichthyes.fasta sequenceData/7-refdb/mitofish.fasta
Output
merging all fasta files and discarding duplicate sequence headers
found 3300 sequences in sequenceData/7-refdb/ncbi16Schondrichthyes.fasta
found 825365 sequences in sequenceData/7-refdb/mitofish.fasta
written 825709 sequences to mergedDB.fasta
2.5 Step 3: extract amplicons through in silico PCR#
From the merged file, we can start with the first curation step of the reference database, i.e., extracting the amplicon sequences. We will be extracting the amplicon region from each sequence through an in silico PCR analysis. With this analysis, we will locate the forward and reverse primer-binding regions and extract the sequence in between. This will significantly reduce file sizes when larger data sets are downloaded, while also making sure that the necessary information is kept.
crabs insilico_pcr -i sequenceData/7-refdb/mergedDB.fasta -o sequenceData/7-refdb/mergedDBinsilico.fasta -f GACCCTATGGAGCTTTAGAC -r CGCTGTTATCCCTADRGTAACT
Output
reading sequenceData/7-refdb/mergedDB.fasta into memory
100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 794191686/794191686 [00:00<00:00, 1114359519.98it/s]
found 825709 sequences in sequenceData/7-refdb/mergedDB.fasta
running in silico PCR on fasta file containing 825709 sequences
counting the number of sequences found by in silico PCR
100%|████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 11323145/11323145 [00:00<00:00, 319601025.34it/s]
found primers in 52758 sequences
reading sequences without primer-binding regions into memory
100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 559830454/559830454 [00:00<00:00, 903207100.93it/s]
reverse complementing 772951 untrimmed sequences
running in silico PCR on 772951 reverse complemented untrimmed sequences
counting the number of sequences found by in silico PCR
100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 78281/78281 [00:00<00:00, 199231985.09it/s]
found primers in 367 sequences
2.6 Step 4: extract amplicons through pairwise global alignments#
Amplicons in the originally downloaded sequences might be missed during the in silico PCR analysis when one or both primer-binding regions are not incorporated in the online deposited sequence. This can happen when the reference barcode is generated with the same primer set or if the deposited sequence is incomplete in the primer-binding regions (denoted as “N” in the sequence). To retrieve those missed amplicons, we can use the already-retrieved amplicons as seed sequences in a Pairwise Global Alignment (PGA) analysis.
crabs pga -i sequenceData/7-refdb/mergedDB.fasta -o sequenceData/7-refdb/mergedDBinsilicoPGA.fasta -db sequenceData/7-refdb/mergedDBinsilico.fasta -f GACCCTATGGAGCTTTAGAC -r CGCTGTTATCCCTADRGTAACT
Output
reading sequenceData/7-refdb/mergedDBinsilico.fasta into memory
95%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████▏ | 10817435/11401426 [00:00<00:00, 100683726.05it/s]
found 53125 number of sequences in sequenceData/7-refdb/mergedDBinsilico.fasta
reading sequenceData/7-refdb/mergedDB.fasta into memory
99%|████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████▌ | 785113607/794191686 [00:05<00:00, 155879553.73it/s]
found 825709 number of sequences in sequenceData/7-refdb/mergedDB.fasta
analysing 772584 number of sequences for pairwise global alignment
100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 558392140/558392140 [00:01<00:00, 321625912.48it/s]
subsetting sequenceData/7-refdb/mergedDB.fasta by discarding sequences longer than 10,000 bp
found 772374 (99.97%) number of sequences in sequenceData/7-refdb/mergedDB.fasta shorter than 10,000 bp
running pairwise global alignment on 772374 number of sequences. This may take a while!
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading file sequenceData/7-refdb/mergedDBinsilico.fasta 100%
10817412 nt in 53124 seqs, min 73, max 1274, avg 204
minseqlength 32: 1 sequence discarded.
Masking 100%
Counting k-mers 100%
Creating k-mer index 100%
Searching 100%
Matching unique query sequences: 2515 of 772374 (0.33%)
filtering alignments based on parameter settings
100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 140747/140747 [00:00<00:00, 11348463.16it/s]
2515 out of 772374 number of sequences aligned to sequenceData/7-refdb/mergedDBinsilico.fasta
1864 number of sequences achieved an alignment that passed filtering thresholds
written 54989 sequences to sequenceData/7-refdb/mergedDBinsilicoPGA.fasta
2.7 Step 5: assigning a taxonomic ID#
The next curating steps for our reference database require a taxonomic ID for each sequence. Currently, sequences are only identified by a unique accession or CRABS number. Therefore, before we continue curating the reference database, we will need to assign a taxonomic lineage to each sequence. The taxonomic ID is based on the NCBI taxonomy, for which we need to download the files. We can do this through the db_download function.
cd sequenceData/7-refdb
crabs db_download -s taxonomy
cd ../../
Output
downloading taxonomy information
nucl_gb.accession2taxid.gz 100%[====================================================================================================>] 2.18G 1.32MB/s in 10m 54s
unzipping nucl_gb.accession2taxid.gz...
taxdump.tar.gz 100%[====================================================================================================>] 60.37M 2.19MB/s in 24s
removing intermediary files
Once the taxonomy files are downloaded, we can assign a taxonomic ID for each amplicon using the assign_tax command.
crabs assign_tax -i sequenceData/7-refdb/mergedDBinsilicoPGA.fasta -o sequenceData/7-refdb/mergedDBinsilicoPGAtax.tsv -a sequenceData/7-refdb/nucl_gb.accession2taxid -t sequenceData/7-refdb/nodes.dmp -n sequenceData/7-refdb/names.dmp -w yes
Output
retrieving accession numbers from sequenceData/7-refdb/mergedDBinsilicoPGA.fasta
95%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████▏ | 11110715/11715190 [00:00<00:00, 103506466.40it/s]
found 54989 accession numbers in sequenceData/7-refdb/mergedDBinsilicoPGA.fasta
reading sequenceData/7-refdb/nucl_gb.accession2taxid into memory
100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 12080791485/12080791485 [01:55<00:00, 104443260.06it/s]
reading sequenceData/7-refdb/nodes.dmp into memory
100%|███████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 191357208/191357208 [00:03<00:00, 48500398.70it/s]
reading sequenceData/7-refdb/names.dmp into memory
100%|███████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 237051517/237051517 [00:03<00:00, 70933792.18it/s]
processed 54961 entries in sequenceData/7-refdb/nucl_gb.accession2taxid
processed 2530548 entries in sequenceData/7-refdb/nodes.dmp
processed 2530548 entries in sequenceData/7-refdb/names.dmp
assigning a tax ID number to 54989 accession numbers from sequenceData/7-refdb/mergedDBinsilicoPGA.fasta
did not find 28 accession numbers in sequenceData/7-refdb/nucl_gb.accession2taxid, retrieving information through a web search
100%|███████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 28/28 [00:45<00:00, 1.63s/it]
could not find a taxonomic ID for 0 entries
54989 accession numbers resulted in 14376 unique tax ID numbers
generating taxonomic lineages for 14376 tax ID numbers
assigning a taxonomic lineage to 54989 accession numbers
written 54989 entries to sequenceData/7-refdb/mergedDBinsilicoPGAtax.tsv
The output file of the assign_tax function has been altered from a 2-line fasta structure to a tab-delimited file whereby all information of a sequence is provided on a single line. Let’s use the head command to investigate the format of the file.
head -n 4 sequenceData/7-refdb/mergedDBinsilicoPGAtax.tsv
Output
OR575552 305515 Eukaryota Chordata Chondrichthyes Squaliformes Dalatiidae Euprotomicrus Euprotomicrus_bispinatus ACTTAAATTAATTATGTAAAATTTTTAACTCTCCGGAGAAAAACCACCCATATAATACCCCTAATTTAACTGTTTTTGGTTGGGGTGACCAGGGGGAAAAAATTATCCCCCCCATCGATTGAGTACTCAGTACTTAAAAATTAGAATGACAACTCTAATTAATAAAACATTTACCGAAACATGACCCAGAATTTATTCTGATCAATGAACCA
OR582714 862652 Eukaryota Chordata Chondrichthyes Squaliformes Etmopteridae Etmopterus Etmopterus_bigelowi ACTTAAATTAATCATATAAACTATTAACCCACGGGAATAAATTACATATATACCCCTAATTTAACTGTTTTTGGTTGGGGCGACCAAGGGGGAGAAAAAATCCCCCTCATCGATTGAGTACTTAGTACTTAAAAATTAGAACGACAGTTCTTATTAATGAAATATTTAACGAAAAATGACCCAGTTTTTCTGATCAATGAACCA
OR582700 263691 Eukaryota Chordata Chondrichthyes Carcharhiniformes Scyliorhinidae Apristurus Apristurus_kampae ACTTAGACTAATTATGTAATTTTTTTCCGCCTGTGGGTAAAAACAAAAATATAATATTTCTAGTTTAATTGTTTTTGGTTGGGGTGACCAAGGGGAAAAACAAATCCCCCTTATCGACCAAGTACTCAGTACTTAAAAATTAGAGCGACAGCTCTAATCAATAAAACATTTATCGAAAAATGACCCAGGATTTCCTGATCAATGAACCA
OR582686 671160 Eukaryota Chordata Chondrichthyes Squaliformes Etmopteridae Etmopterus Etmopterus_gracilispinis ACTTAAATTAATTATGTAAAACTACTAATCCACGGAAATAAACTATTTATATAATATTTCTAATTTAACTGTTTTTGGTTGGGGTGACCGAGGGGAAAAGAAAATCCCCCTCATCGATTGAGTACTTAGTACTTAAAAATTAGAACGACAGTTCTTATTAATAAAATATTTAACGAAAAATGACCCAGTTTTTCTGATCAATGAACCA
2.8 Step 6: dereplicating the DB#
Now that we have a taxonomic ID for each amplicon sequence in our reference database, we can remove duplicate sequences per species, i.e., dereplicating the reference database.
crabs dereplicate -i sequenceData/7-refdb/mergedDBinsilicoPGAtax.tsv -o sequenceData/7-refdb/mergedDBinsilicoPGAtaxderep.tsv -m uniq_species
Output
dereplicating sequenceData/7-refdb/mergedDBinsilicoPGAtax.tsv, keeping all unique sequences per species
species information found at position 7, starting dereplication process
100%|████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 16890510/16890510 [00:00<00:00, 201942731.88it/s]
found 54989 sequences in sequenceData/7-refdb/mergedDBinsilicoPGAtax.tsv
written 25135 sequences to sequenceData/7-refdb/mergedDBinsilicoPGAtaxderep.tsv
2.8 Step 7: ref DB cleanup#
The final curation step for our custom reference database is to clean up the database using a variety of parameters, including minimum length, maximum length, maximum number of ambiguous base calls, environmental sequences, sequences for which the species name is not provided, and sequences with unspecified taxonomic levels.
crabs seq_cleanup -i sequenceData/7-refdb/mergedDBinsilicoPGAtaxderep.tsv -o sequenceData/7-refdb/mergedDBinsilicoPGAtaxderepclean.tsv -e yes -s yes -na 1
Output
CRABS v0.1.8
https://github.com/gjeunen/reference_database_creator
Filter INPUT: 100.0%|███████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████|00:00<00:00
SUMMARY STATISTICS:
Number of sequences analysed: 25135
Sequences kept: 22121 (88.01%)
Sequences removed: 3014 (11.99%)
Too short sequences: 326
Too long sequences: 12
Sequences with too many "N": 622
Environmental sequences: 0
Sequences without proper species ID: 2099
Sequences with too many undefined taxonomic levels: 3
2.9 Step 8: export ref DB#
Now that we have created a custom curated reference database containing 22,121 16S rRNA gene sequences of fish and sharks, we can export the reference database to be used by different taxonomy classifiers. Multiple formats are implemented in CRABS. Today, we will export the reference database in SINTAX format, as this is one of the classification methods we will be using later on with our COI data set.
crabs tax_format -i sequenceData/7-refdb/mergedDBinsilicoPGAtaxderepclean.tsv -o sequenceData/7-refdb/mergedDBinsilicoPGAtaxderepcleansintax.fasta -f sintax
head -n 4 sequenceData/7-refdb/mergedDBinsilicoPGAtaxderepcleansintax.fasta
Output
>OR575552;tax=d:Eukaryota,p:Chordata,c:Chondrichthyes,o:Squaliformes,f:Dalatiidae,g:Euprotomicrus,s:Euprotomicrus_bispinatus
ACTTAAATTAATTATGTAAAATTTTTAACTCTCCGGAGAAAAACCACCCATATAATACCCCTAATTTAACTGTTTTTGGTTGGGGTGACCAGGGGGAAAAAATTATCCCCCCCATCGATTGAGTACTCAGTACTTAAAAATTAGAATGACAACTCTAATTAATAAAACATTTACCGAAACATGACCCAGAATTTATTCTGATCAATGAACCA
>OR582714;tax=d:Eukaryota,p:Chordata,c:Chondrichthyes,o:Squaliformes,f:Etmopteridae,g:Etmopterus,s:Etmopterus_bigelowi
ACTTAAATTAATCATATAAACTATTAACCCACGGGAATAAATTACATATATACCCCTAATTTAACTGTTTTTGGTTGGGGCGACCAAGGGGGAGAAAAAATCCCCCTCATCGATTGAGTACTTAGTACTTAAAAATTAGAACGACAGTTCTTATTAATGAAATATTTAACGAAAAATGACCCAGTTTTTCTGATCAATGAACCA
3. The COI reference DB#
For our tutorial data set, we will be using the COI reference database: sequenceData/7-refdb/newDBcrabsCOIsintax.fasta, which I made through CRABS, supplemented with local barcodes provided by HKU, and subsetted to reduce the file size and speed up classification by excluding non-target taxonomic groups such as land plants and insects for example. This reference database is formatted according to SINTAX specifications and holds 213,937 high quality, unique, COI reference barcodes.
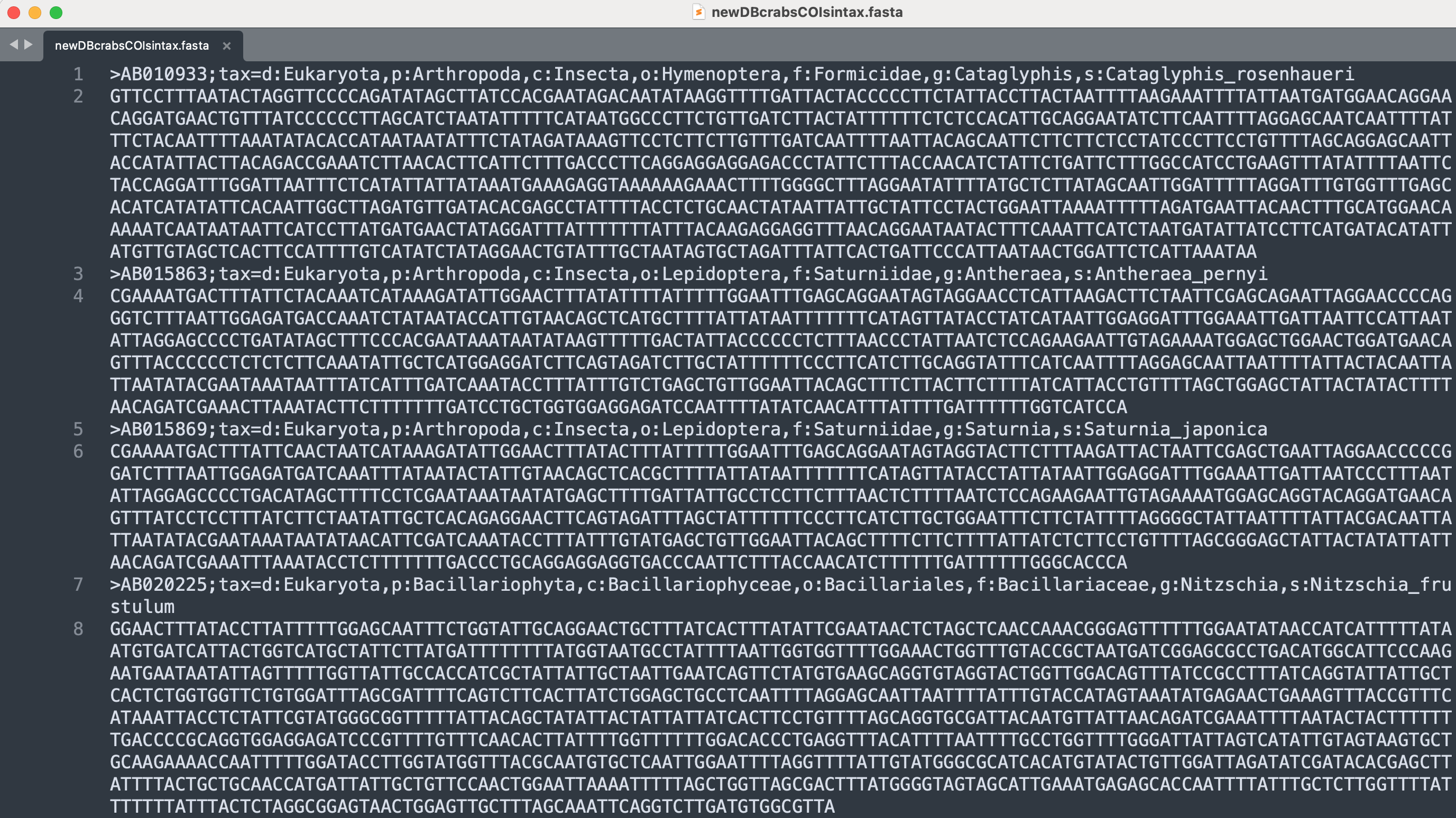
Fig. 23 : A screenshot of the COI reference database#
4. Taxonomic classification#
4.1 SINTAX#
Since the COI reference database sequenceData/7-refdb/newDBcrabsCOIsintax.fasta is already formatted according to SINTAX specifications, let us first explore the SINTAX algorithm. The SINTAX algorithm predicts taxonomy by using k-mer similarity to identify the top hit in a reference database and provides bootstrap confidence for all ranks in the prediction. Unlike the RDP classifier, SINTAX does not rely on Bayesian posteriors. Hence, training the data set is not necessary and results can be obtained very quickly. SINTAX was developed for USEARCH and is also incorporated into VSEARCH through the --sintax command. More information about the SINTAX algorithm can be found on the USEARCH website.
The input file for the --sintax command is our ZOTU sequence list sequenceData/8-final/zotus.fasta. The database to match our sequences can be specified using the --db parameter. Finally, the output file is a simple tab-delimited text file, which can be specified using the --tabbedout parameter. Although not essential, you can provide a confidence value cutoff level through the --sintax_cutoff parameter. For the tutorial data set, let’s set the similarity threshold (--sintax_cutoff) at 60%.
vsearch --sintax sequenceData/8-final/zotus.fasta --db sequenceData/7-refdb/newDBcrabsCOIsintax.fasta --tabbedout sequenceData/8-final/sintaxTaxonomy.txt --sintax_cutoff 0.60
Output
vsearch v2.23.0_macos_aarch64, 16.0GB RAM, 8 cores
https://github.com/torognes/vsearch
Reading file sequenceData/7-refdb/newDBcrabsCOIsintax.fasta 100%
87166478 nt in 213930 seqs, min 101, max 47038, avg 407
maxseqlength 50000: 7 sequences discarded.
Counting k-mers 100%
Creating k-mer index 100%
Classifying sequences 100%
Classified 1345 of 1345 sequences (100.00%)
The output shows that SINTAX managed to classify all 1,345 ZOTU sequences. Using the head command, we can inspect the first couple of lines in our document.
head -n 10 sequenceData/8-final/sintaxTaxonomy.txt
Output
zotu.6 d:Eukaryota(0.98),p:Bacillariophyta(0.38),c:Fragilariophyceae(0.07),o:Licmophorales(0.07),f:Ulnariaceae(0.07),g:Ulnaria(0.07),s:Ulnaria_acus(0.07) + d:Eukaryota
zotu.3 d:Eukaryota(1.00),p:Arthropoda(1.00),c:Malacostraca(1.00),o:Decapoda(1.00),f:Alpheidae(1.00),g:Athanas(1.00),s:parvus(1.00) + d:Eukaryota,p:Arthropoda,c:Malacostraca,o:Decapoda,f:Alpheidae,g:Athanas,s:parvus
zotu.1 d:Eukaryota(0.98),p:Bacillariophyta(0.17),c:Fragilariophyceae(0.05),o:Licmophorales(0.05),f:Ulnariaceae(0.05),g:Ulnaria(0.05),s:Ulnaria_acus(0.05) + d:Eukaryota
zotu.8 d:Eukaryota(1.00),p:Annelida(1.00),c:Polychaeta(1.00),o:Phyllodocida(1.00),f:Nereididae(1.00),g:Platynereis(1.00),s:HK01(1.00) + d:Eukaryota,p:Annelida,c:Polychaeta,o:Phyllodocida,f:Nereididae,g:Platynereis,s:HK01
zotu.2 d:Eukaryota(1.00),p:Bacillariophyta(0.13),c:Coscinodiscophyceae(0.02),o:Thalassiosirales(0.02),f:Thalassiosiraceae(0.02),g:Thalassiosira(0.02),s:Thalassiosira_nordenskioeldii(0.02d:Eukaryota
zotu.11 d:Eukaryota(1.00),p:Bacillariophyta(0.33),c:Bacillariophyceae(0.21),o:Bacillariales(0.17),f:Bacillariaceae(0.17),g:Cylindrotheca(0.07),s:Cylindrotheca_closterium(0.07) + d:Eukaryota
zotu.4 d:Eukaryota(1.00),p:Arthropoda(1.00),c:Malacostraca(1.00),o:Decapoda(1.00),f:Decapoda_nan(0.96),g:Decapoda_nan_nan(0.96),s:Caridea_sp.(0.96) + d:Eukaryota,p:Arthropoda,c:Malacostraca,o:Decapoda,f:Decapoda_nan,g:Decapoda_nan_nan,s:Caridea_sp.
zotu.5 d:Eukaryota(0.99),p:Chordata(0.17),c:Actinopteri(0.08),o:Perciformes(0.04),f:Serranidae(0.01),g:Aporops(0.01),s:Aporops_bilinearis(0.01) + d:Eukaryota
zotu.10 d:Eukaryota(1.00),p:Arthropoda(1.00),c:Malacostraca(1.00),o:Decapoda(1.00),f:Rhynchocinetidae(1.00),g:Rhynchocinetes(1.00),s:brucei(1.00) + d:Eukaryota,p:Arthropoda,c:Malacostraca,o:Decapoda,f:Rhynchocinetidae,g:Rhynchocinetes,s:brucei
zotu.7 d:Eukaryota(1.00),p:Bacillariophyta(0.22),c:Mediophyceae(0.02),o:Cymatosirales(0.02),f:Cymatosiraceae(0.02),g:Minutocellus(0.02),s:Minutocellus_polymorphus(0.02) + d:Eukaryota
The head command output shows that our file is structured in 4 columns, the first containing the ZOTU sequence name, the second providing the classification information without the cut off threshold, the third specifying on which strand the match was made, and the fourth column providing the classification information with the cut off value of 60% applied. From the head command, we can also see that the ZOTU sequence list is not ordered based on sequence number. Something you might want to keep in mind when you want to parse the document.
4.2 IDTAXA#
The second method we will explore is the IDTAXA machine learning algorithm. The IDTAXA algorithm is a machine learning algorithm that is split into two phases: a “training” phase where the classifier learns attributes of the training set, and a “testing” phase where sequences with unknown taxonomic assignments are classified. The IDTAXA algorithm is incorporated in the DECIPHER R package.
4.2.1 Input formatting#
IDTAXA requires three files to run, including (i) a reference database containing sequences with known taxonomic IDs, (ii) a taxonomic ID file containing information about taxonomic ranks, and (iii) the ZOTU sequence file which we created during the bioinformatic processing of our sequence data. We already have access to the third ZOTU file (sequenceData/8-final/zotus.fasta) and we can create the first tow files from the SINTAX reference database file (sequenceData/7-refdb/newDBcrabsCOIsintax.fasta). The IDTAXA reference database is a fasta file with an altered header structure compared to the SINTAX format we used above. The taxonomic ID file is a simple text file containing five columns separated with an asterisk (’*’). Column 1 refers to the unique index number of to taxon name, column 2 refers to the taxon name, column 3 reference to the unique index number of the parent, column 4 represents the numerical value of the taxonomic level, and column 5 refers to the name of the taxonomic rank.
It should be noted that the IDTAXA algorithm, as implemented in DECIPHER, cannot resolve homonyms, i.e., identical taxon names. IDTAXA will error out when encountering homonyms. To solve this issue, we will create the reference database file and taxonomic ID file using names of two ranks pasted together to generate unique names. For example, the fish family name of Chilodontidae will be transformed to the order name + the family name: CharaciformesChilodontidae, while the gastropod family name of Chilodontidae will be transformed to SeguenziidaChilodontidae. This will be done for every single name, no matter if they are unique or a homonym exists. This “hack” is also the reason why the taxonomic names in the output file will follow this structure. Don’t worry about the formatting of the output file. We will correct the taxonomic names during the file parsing process in section 5.2 IDTAXA formatting.
To format the sequenceData/7-refdb/newDBcrabsCOIsintax.fasta file to IDTAXA format, we will execute the python script below. This script takes in three user arguments, including the SINTAX reference database, the output file name for the IDTAXA reference database, and the output file name for the IDTAXA taxonomic ID text file.
nano sequenceData/1-scripts/idtaxaInitFormat
#! /usr/bin/env python3
import sys
inFile = sys.argv[1]
dbOut = sys.argv[2]
taxOut = sys.argv[3]
indexNameDict = {'Root' : 0}
parentDict = {'Root' : -1}
levelDict = {'Root' : 0}
rankDict = {'Root' : 'rootrank'}
indexNumber = 0
ranks = {0: 'rootrank', 1 : 'domain', 2 : 'phylum', 3 : 'class', 4 : 'order', 5: 'family', 6 : 'genus', 7 : 'species'}
idTaxaSeqDict = {}
with open(inFile, 'r') as infile:
for line in infile:
previousName = 'Root'
previousTaxonName = 'Root'
oldName = 'start'
levelTax = 0
if line.startswith('>CRABS'):
line = line.rstrip('\n')
seqID = line.split(';')[0]
headerStart = f'{seqID};Root'
lineage = line.split(':')[2:9]
for taxon in lineage:
levelTax += 1
taxonName = taxon.split(',')[0]
if taxonName == '':
taxonName = f'{previousName}_insertaeCedis'
uniqueTaxonName = f'{previousTaxonName}{taxonName}'
headerStart = f'{headerStart};{uniqueTaxonName}'
if uniqueTaxonName not in indexNameDict:
indexNumber += 1
indexNameDict[uniqueTaxonName] = indexNumber
parentDict[uniqueTaxonName] = indexNameDict[previousName]
levelDict[uniqueTaxonName] = levelTax
rankDict[uniqueTaxonName] = ranks[levelTax]
taxonNumber = indexNameDict[uniqueTaxonName]
oldName = previousName
previousName = uniqueTaxonName
previousTaxonName = taxonName
elif line.startswith('>'):
line = line.rstrip('\n')
seqID = line.split(';')[0]
headerStart = f'{seqID};Root'
lineage = line.split(':')[1:8]
for taxon in lineage:
levelTax += 1
taxonName = taxon.split(',')[0]
if taxonName == '':
taxonName = f'{previousName}_insertaeCedis'
uniqueTaxonName = f'{previousTaxonName}{taxonName}'
headerStart = f'{headerStart};{uniqueTaxonName}'
if uniqueTaxonName not in indexNameDict:
indexNumber += 1
indexNameDict[uniqueTaxonName] = indexNumber
parentDict[uniqueTaxonName] = indexNameDict[previousName]
levelDict[uniqueTaxonName] = levelTax
rankDict[uniqueTaxonName] = ranks[levelTax]
taxonNumber = indexNameDict[uniqueTaxonName]
oldName = previousName
previousName = uniqueTaxonName
previousTaxonName = taxonName
else:
idTaxaSeqDict[headerStart] = line.rstrip('\n')
with open(taxOut, 'w') as outfile:
for indexName in indexNameDict:
printLine = f'{indexNameDict[indexName]}*{indexName}*{parentDict[indexName]}*{levelDict[indexName]}*{rankDict[indexName]}\n'
_ = outfile.write(printLine)
with open(dbOut, 'w') as seqFile:
for key in idTaxaSeqDict:
_ = seqFile.write(f'{key}\n{idTaxaSeqDict[key]}\n')
Press ctrl + x to exit out of the editor, followed by y and return.
chmod +x sequenceData/1-scripts/idtaxaInitFormat
./sequenceData/1-scripts/idtaxaInitFormat sequenceData/7-refdb/newDBcrabsCOIsintax.fasta sequenceData/7-refdb/idtaxa.fasta sequenceData/7-refdb/idtaxa.txt
Let’s check if the two IDTAXA files have been created in the sequenceData/7-refdb/ subdirectory and explore their structure using the head command.
ls -ltr sequenceData/7-refdb
head -n 10 sequenceData/7-refdb/idtaxa.fasta
head -n 10 sequenceData/7-refdb/idtaxa.txt
Output
-rwxr-xr-x@ 1 gjeunen staff 240602642 6 Oct 20:28 newDBcrabsCOIsintax.fasta
-rw-r--r-- 1 gjeunen staff 7098352 7 Oct 20:42 idtaxa.txt
-rw-r--r-- 1 gjeunen staff 268731720 7 Oct 20:42 idtaxa.fasta
>AB010933;Root;RootEukaryota;EukaryotaArthropoda;ArthropodaInsecta;InsectaHymenoptera;HymenopteraFormicidae;FormicidaeCataglyphis;CataglyphisCataglyphis_rosenhaueri
GTTCCTTTAATACTAGGTTCCCCAGATATAGCTTATCCACGAATAGACAATATAAGGTTTTGATTACTACCCCCTTCTATTACCTTACTAATTTTAAGAAATTTTATTAATGATGGAACAGGAACAGGATGAACTGTTTATCCCCCCTTAGCATCTAATATTTTTCATAATGGCCCTTCTGTTGATCTTACTATTTTTTCTCTCCACATTGCAGGAATATCTTCAATTTTAGGAGCAATCAATTTTATTTCTACAATTTTAAATATACACCATAATAATATTTCTATAGATAAAGTTCCTCTTCTTGTTTGATCAATTTTAATTACAGCAATTCTTCTTCTCCTATCCCTTCCTGTTTTAGCAGGAGCAATTACCATATTACTTACAGACCGAAATCTTAACACTTCATTCTTTGACCCTTCAGGAGGAGGAGACCCTATTCTTTACCAACATCTATTCTGATTCTTTGGCCATCCTGAAGTTTATATTTTAATTCTACCAGGATTTGGATTAATTTCTCATATTATTATAAATGAAAGAGGTAAAAAAGAAACTTTTGGGGCTTTAGGAATATTTTATGCTCTTATAGCAATTGGATTTTTAGGATTTGTGGTTTGAGCACATCATATATTCACAATTGGCTTAGATGTTGATACACGAGCCTATTTTACCTCTGCAACTATAATTATTGCTATTCCTACTGGAATTAAAATTTTTAGATGAATTACAACTTTGCATGGAACAAAAATCAATAATAATTCATCCTTATGATGAACTATAGGATTTATTTTTTTATTTACAAGAGGAGGTTTAACAGGAATAATACTTTCAAATTCATCTAATGATATTATCCTTCATGATACATATTATGTTGTAGCTCACTTCCATTTTGTCATATCTATAGGAACTGTATTTGCTAATAGTGCTAGATTTATTCACTGATTCCCATTAATAACTGGATTCTCATTAAATAA
>AB015863;Root;RootEukaryota;EukaryotaArthropoda;ArthropodaInsecta;InsectaLepidoptera;LepidopteraSaturniidae;SaturniidaeAntheraea;AntheraeaAntheraea_pernyi
CGAAAATGACTTTATTCTACAAATCATAAAGATATTGGAACTTTATATTTTATTTTTGGAATTTGAGCAGGAATAGTAGGAACCTCATTAAGACTTCTAATTCGAGCAGAATTAGGAACCCCAGGGTCTTTAATTGGAGATGACCAAATCTATAATACCATTGTAACAGCTCATGCTTTTATTATAATTTTTTTCATAGTTATACCTATCATAATTGGAGGATTTGGAAATTGATTAATTCCATTAATATTAGGAGCCCCTGATATAGCTTTCCCACGAATAAATAATATAAGTTTTTGACTATTACCCCCCTCTTTAACCCTATTAATCTCCAGAAGAATTGTAGAAAATGGAGCTGGAACTGGATGAACAGTTTACCCCCCTCTCTCTTCAAATATTGCTCATGGAGGATCTTCAGTAGATCTTGCTATTTTTTCCCTTCATCTTGCAGGTATTTCATCAATTTTAGGAGCAATTAATTTTATTACTACAATTATTAATATACGAATAAATAATTTATCATTTGATCAAATACCTTTATTTGTCTGAGCTGTTGGAATTACAGCTTTCTTACTTCTTTTATCATTACCTGTTTTAGCTGGAGCTATTACTATACTTTTAACAGATCGAAACTTAAATACTTCTTTTTTTGATCCTGCTGGTGGAGGAGATCCAATTTTATATCAACATTTATTTTGATTTTTTGGTCATCCA
>AB015869;Root;RootEukaryota;EukaryotaArthropoda;ArthropodaInsecta;InsectaLepidoptera;LepidopteraSaturniidae;SaturniidaeSaturnia;SaturniaSaturnia_japonica
CGAAAATGACTTTATTCAACTAATCATAAAGATATTGGAACTTTATACTTTATTTTTGGAATTTGAGCAGGAATAGTAGGTACTTCTTTAAGATTACTAATTCGAGCTGAATTAGGAACCCCCGGATCTTTAATTGGAGATGATCAAATTTATAATACTATTGTAACAGCTCACGCTTTTATTATAATTTTTTTCATAGTTATACCTATTATAATTGGAGGATTTGGAAATTGATTAATCCCTTTAATATTAGGAGCCCCTGACATAGCTTTTCCTCGAATAAATAATATGAGCTTTTGATTATTGCCTCCTTCTTTAACTCTTTTAATCTCCAGAAGAATTGTAGAAAATGGAGCAGGTACAGGATGAACAGTTTATCCTCCTTTATCTTCTAATATTGCTCACAGAGGAACTTCAGTAGATTTAGCTATTTTTTCCCTTCATCTTGCTGGAATTTCTTCTATTTTAGGGGCTATTAATTTTATTACGACAATTATTAATATACGAATAAATAATATAACATTCGATCAAATACCTTTATTTGTATGAGCTGTTGGAATTACAGCTTTTCTTCTTTTATTATCTCTTCCTGTTTTAGCGGGAGCTATTACTATATTATTAACAGATCGAAATTTAAATACCTCTTTTTTTGACCCTGCAGGAGGAGGTGACCCAATTCTTTACCAACATCTTTTTTGATTTTTTGGGCACCCA
>AB020225;Root;RootEukaryota;EukaryotaBacillariophyta;BacillariophytaBacillariophyceae;BacillariophyceaeBacillariales;BacillarialesBacillariaceae;BacillariaceaeNitzschia;NitzschiaNitzschia_frustulum
TTTATCAGGTATTATTGCTCACTCTGGTGGTTCTGTGGATTTAGCGATTTTCAGTCTTCACTTATCTGGAGCTGCCTCAATTTTAGGAGCAATTAATTTTATTTGTACCATAGTAAATATGAGAACTGAAAGTTTACCGTTTCATAAATTACCTCTATTCGTATGGGCGGTTTTTATTACAGCTATATTACTATTATTATCACTTCCTGTTTTAGCAGGTGCGATTACAATGTTATTAACAGATCGAAATTTTAATACTACTTTTTTTGACCCCGCAGGTGGAGGAGATCCCGTTTTGTTTCAACACTTATTT
>AB020228;Root;RootEukaryota;EukaryotaBacillariophyta;BacillariophytaFragilariophyceae;FragilariophyceaeThalassionemales;ThalassionemalesThalassionemataceae;ThalassionemataceaeThalassionema;ThalassionemaThalassionema_nitzschioides
ACTATCAAGTATAATCGCTCATTCTGGCGGTTCGGTAGATTTAGCAATTTTTAGTTTACATGTTTCTGGAGCGGCTTCAATTTTAGGAGCAATTAATTTTATTTGTACTATTTTTAACATGCGTGTAAAAAGTTTATCATTTCATAAATTACCATTATTTGTTTGGGCTATTTTAATAACTGCCGTTTTACTTCTTTTATCTTTACCTGTTTTAGCAGGGGCAATTACAATGTTATTAACTGATAGAAATTTTAATACAACTTTTTTTGATCCAGCAGGAGGAGGTGATCCGGTGTTATATCAACATTTATTT
0*Root*-1*0*rootrank
1*RootEukaryota*0*1*domain
2*EukaryotaArthropoda*1*2*phylum
3*ArthropodaInsecta*2*3*class
4*InsectaHymenoptera*3*4*order
5*HymenopteraFormicidae*4*5*family
6*FormicidaeCataglyphis*5*6*genus
7*CataglyphisCataglyphis_rosenhaueri*6*7*species
8*InsectaLepidoptera*3*4*order
9*LepidopteraSaturniidae*8*5*family
The output of the head commands shows the structure of both files. Note the slight difference in structure between the IDTAXA and SINTAX reference database format. We can also see the “hack” applied to sequenceData/7-refdb-idtaxa.txt, whereby the names are pasted together, e.g., RootEukaryota, rather than Eukaryota.
4.2.2 Classifier training#
Once the reference database has been formatted to IDTAXA format, we need to train the classifier on the training set, i.e., the formatted reference database. Since IDTAXA is incorporated into the R package DECIPHER, the following code needs to be run in an R environment, such as RStudio. I suggest opening a new R script to copy-paste all the remaining code for this section and save it as sequenceData/1-scripts/idtaxa.R.
library(DECIPHER)
setwd('/Users/gjeunen/Documents/work/lectures/HKU2023/tutorial/sequenceData/7-refdb')
set.seed(1989)
seqIDTAXA <- readDNAStringSet('idtaxa.fasta')
rankIDTAXA <- read.table('idtaxa.txt', header = FALSE, col.names = c('Index', 'Name', 'Parent', 'Level', 'Rank'), sep = '*', quote = '', stringsAsFactors = FALSE)
groups <- names(seqIDTAXA)
head(groups)
groups <- gsub("(.*)(Root;)", "\\2", groups)
head(groups)
groupCounts <- table(groups)
uniqueGroups <- names(groupCounts)
length(uniqueGroups)
# count number of sequences per group and, optionally, select only a subset of sequences if the group is deemed too large (10 seqs)
maxGroupSize <- 10 # max sequences per label (>= 1)
remove <- logical(length(seqIDTAXA))
for (i in which(groupCounts > maxGroupSize)) {
index <- which(groups==uniqueGroups[i])
keep <- sample(length(index),
maxGroupSize)
remove[index[-keep]] <- TRUE
}
sum(remove) # number of sequences eliminated
# train the classifier. Set 'maxIterations' to number of times the classifier will be trained.
maxIterations <- 3 # must be >= 1
allowGroupRemoval <- FALSE
probSeqsPrev <- integer() # suspected problem sequences from prior iteration
for (i in seq_len(maxIterations)) {
cat("Training iteration: ", i, "\n", sep="")
# train the classifier
trainingSet <- LearnTaxa(seqIDTAXA[!remove],
names(seqIDTAXA)[!remove],
rankIDTAXA, maxChildren = 10)
# look for problem sequences
probSeqs <- trainingSet$problemSequences$Index
if (length(probSeqs)==0) {
cat("No problem sequences remaining.\n")
break
} else if (length(probSeqs)==length(probSeqsPrev) &&
all(probSeqsPrev==probSeqs)) {
cat("Iterations converged.\n")
break
}
if (i==maxIterations)
break
probSeqsPrev <- probSeqs
# remove any problem sequences
index <- which(!remove)[probSeqs]
remove[index] <- TRUE # remove all problem sequences
if (!allowGroupRemoval) {
# replace any removed groups
missing <- !(uniqueGroups %in% groups[!remove])
missing <- uniqueGroups[missing]
if (length(missing) > 0) {
index <- index[groups[index] %in% missing]
remove[index] <- FALSE # don't remove
}
}
}
sum(remove)
length(probSeqs)
trainingSet
plot(trainingSet)
Output
> trainingSet
A training set of class 'Taxa'
* K-mer size: 9
* Number of rank levels: 9
* Total number of sequences: 134730
* Number of groups: 44133
* Number of problem groups: 0
* Number of problem sequences: 8
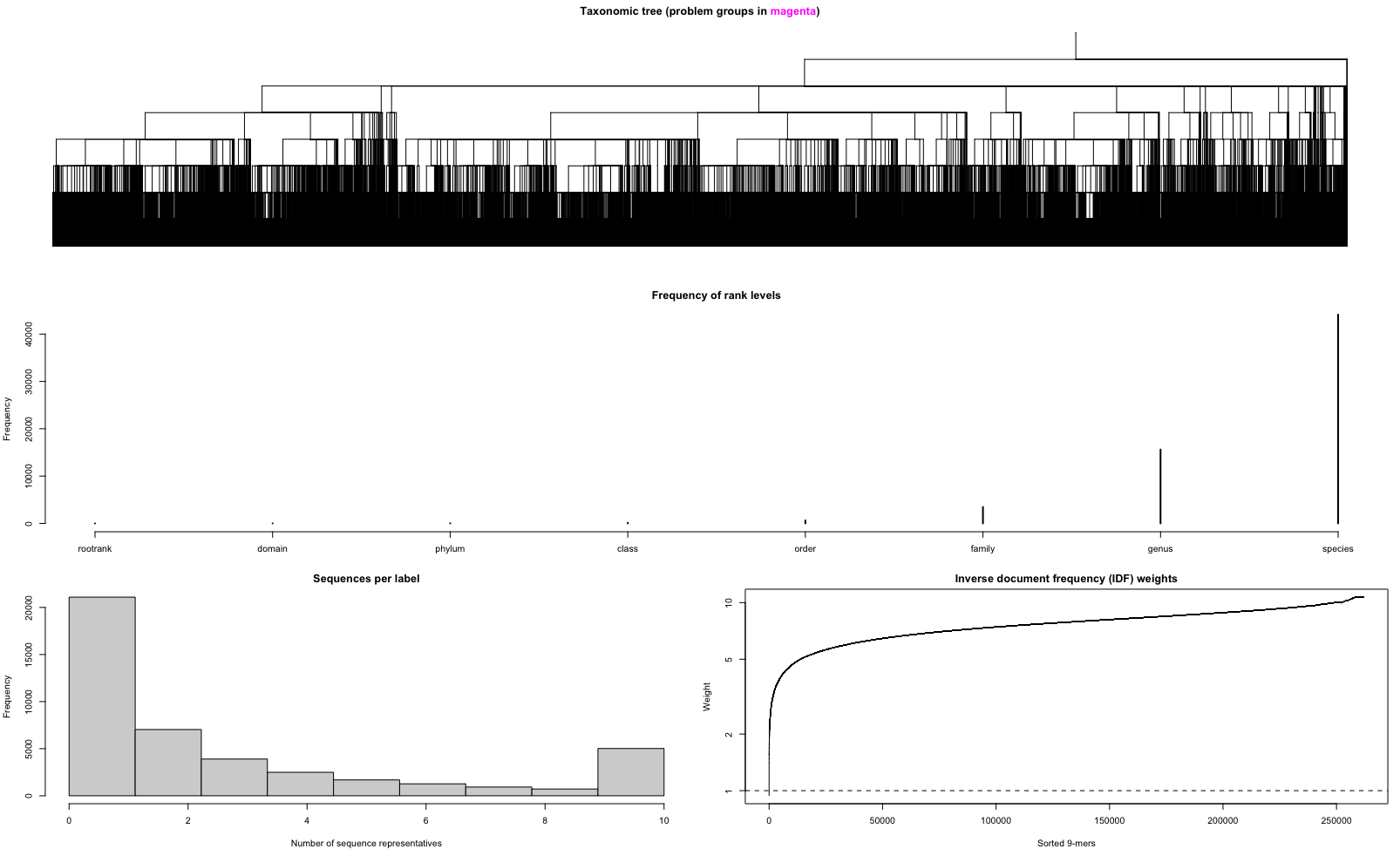
Fig. 24 : The plot of the IDTAXA training set.#
The output of the trainingSet shows that the reference database was trained on a k-mer size of 9 bp, we have 9 ranks incorporated in the taxonomic lineage, a total number of 134,730 sequences, and a total number of 44,133 taxonomic groups. This sequence number is lower than previously mentioned. During the training stage of the classifier, only 10 reads per species were kept (as recommended by the developers), even though CRABS identified them as being unique for that species. For this trained classifier, we iterated over the reference database 3 times. After three iterations, 8 sequences are identified as problematic, i.e., potentially misclassified. Those are represented as magenta lines in the taxonomic tree figure. The figures below provide additional information on the number of sequences with a species ID and a bar graph showing the number of sequences per species.
4.2.3 Classifier training#
Now that the classifier is trained on the reference barcodes, we can use the training set to assign taxonomic IDs to new sequences, i.e., our ZOTU sequences. So, let’s load our ZOTU fasta file and assign taxonomic IDs using the IDTAXA algorithm. Again, the code below should be run in an R environment, such as RStudio. Copy-paste the R code below after the R code used to load in the training set.
Set seed for reproducibility
The IDTAXA algorithm uses bootstrapping, which involves random sampling to obtain a confidence score. For this reason, the classifications are expected to change slightly if the classification process is repeated with the same inputs. To allow for the replicability of the results, we can set a random seed number before classification using the command set.seed(1989). You can find this line of code in the R-script above, when we loaded in the training set. In case this is undesirable for your intent, remove this specific line from the R code to allow for randomness of subsampling during bootstrapping.
# read in the ZOTU fasta file
fastaIDTAXA <- readDNAStringSet('../8-final/zotus.fasta')
# classify sequences
ids <- IdTaxa(fastaIDTAXA, trainingSet, type = 'extended', strand = 'top', threshold = 40, processors = NULL)
ids
# plot results
plot(ids, trainingSet)
# export output
output <- sapply(ids,
function(id) {
paste(id$taxon,
" (",
round(id$confidence, digits = 1),
"%)",
sep = "",
collapse = ": ")
})
# Create an empty character vector to store the output
outputFile <- character(0)
# Combine the zotu ID and information and add them to the output vector
for (zotu_id in names(output)) {
entry <- paste(zotu_id, output[zotu_id], sep = "\t") # Use tab as a separator
outputFile <- c(outputFile, entry)
}
# Write output to file
writeLines(outputFile, '../8-final/idtaxaTaxonomy.txt')
Output
> ids <- IdTaxa(fastaIDTAXA, trainingSet, type = 'extended', strand = 'top', threshold = 40, processors = NULL)
|===============================================================================================================| 100%
Time difference of 507.63 secs
> ids
A test set of class 'Taxa' with length 1345
confidence name taxon
[1] 41% zotu.1 Root; RootEukaryota; unclassified_RootEukaryota...
[2] 43% zotu.2 Root; RootEukaryota; unclassified_RootEukaryota...
[3] 95% zotu.3 Root; RootEukaryota; EukaryotaArthropoda; ArthropodaMalacostraca; MalacostracaD...
[4] 51% zotu.4 Root; RootEukaryota; EukaryotaArthropoda; ArthropodaMalacostraca; MalacostracaD...
[5] 47% zotu.5 Root; RootEukaryota; unclassified_RootEukaryota...
... ... ... ...
[1341] 47% zotu.1341 Root; RootEukaryota; unclassified_RootEukaryota
[1342] 43% zotu.1342 Root; RootEukaryota; unclassified_RootEukaryota
[1343] 56% zotu.1343 Root; RootEukaryota; unclassified_RootEukaryota
[1344] 53% zotu.1344 Root; RootEukaryota; unclassified_RootEukaryota
[1345] 55% zotu.1345 Root; RootEukaryota; unclassified_RootEukaryota
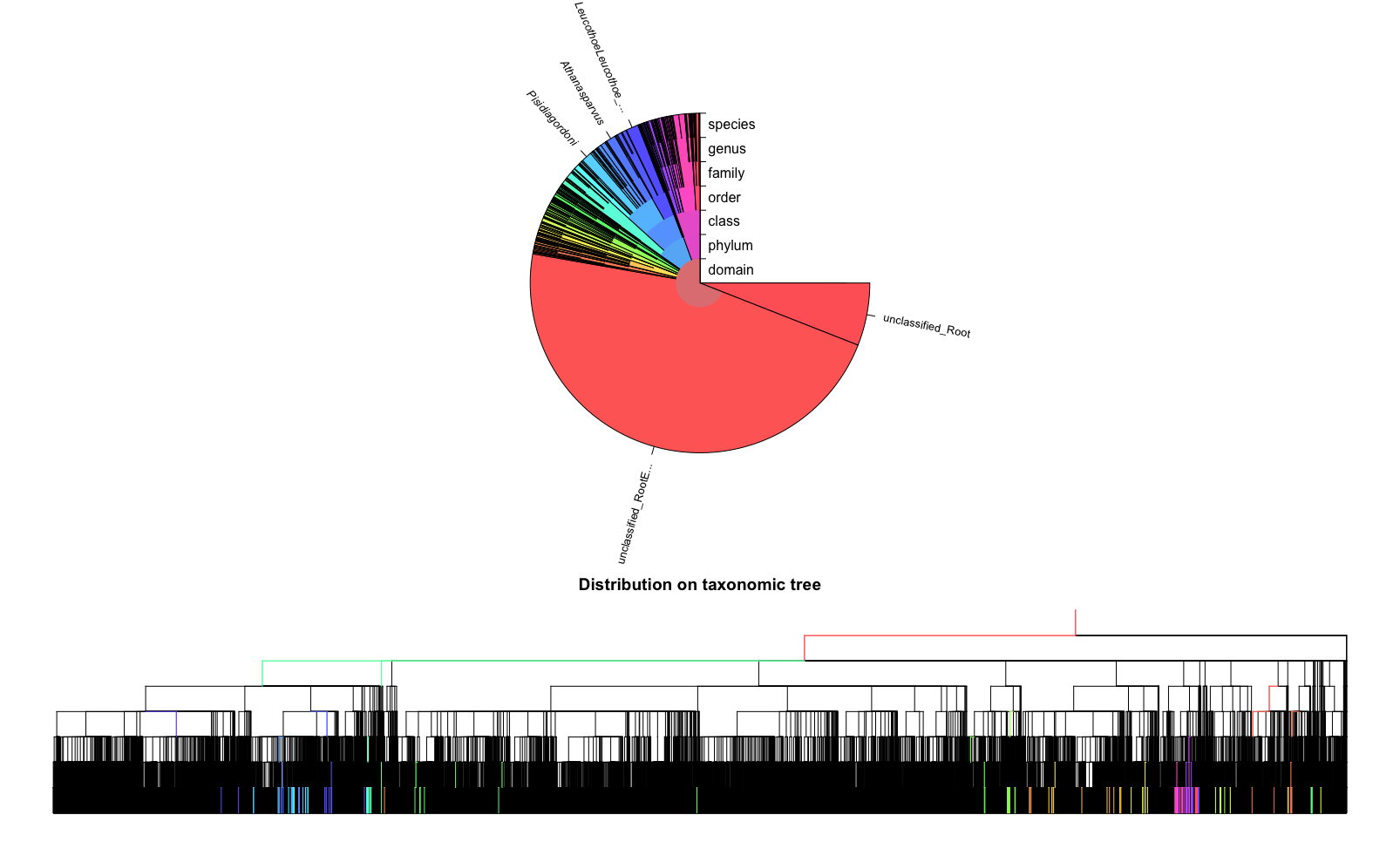
Fig. 25 : The plot of the IDTAXA results.#
Plotting the IDTAXA results shows a pie graph with all the different taxonomic groups coloured differently, as well as a taxonomic tree where coloured branches (same as the pie graph colours) represent the reference barcodes that were matched to the query sequences. Note the difference in time it took between SINTAX and IDTAXA. Also note that IDTAXA classifies a large proportion to Eukaryota, as the authors describe IDTAXA as a more conserved classifier compared to other implementations.
We also exported the results to sequenceData/8-final/idtaxaTaxonomy.txt. Let us look at the structure of this file using the head command.
head -n 10 sequenceData/8-final/idtaxaTaxonomy.txt
Output
zotu.1 Root (41.2%): RootEukaryota (41.2%): unclassified_RootEukaryota (41.2%)
zotu.2 Root (43.9%): RootEukaryota (43.9%): unclassified_RootEukaryota (43.9%)
zotu.3 Root (95.2%): RootEukaryota (95.2%): EukaryotaArthropoda (95.2%): ArthropodaMalacostraca (95.2%): MalacostracaDecapoda (95.2%): DecapodaAlpheidae (95.2%): AlpheidaeAthanas (95.2%): Athanasparvus (95.2%)
zotu.4 Root (100%): RootEukaryota (100%): EukaryotaArthropoda (100%): ArthropodaMalacostraca (100%): MalacostracaDecapoda (100%): DecapodaHippolytidae (100%): HippolytidaeThor (95.7%): ThorHK01 (51.5%)
zotu.5 Root (47.9%): RootEukaryota (47.6%): unclassified_RootEukaryota (47.6%)
zotu.6 Root (44.1%): RootEukaryota (43.7%): unclassified_RootEukaryota (43.7%)
zotu.7 Root (42.6%): RootEukaryota (42.6%): unclassified_RootEukaryota (42.6%)
zotu.8 Root (100%): RootEukaryota (100%): EukaryotaAnnelida (100%): AnnelidaPolychaeta (100%): PolychaetaPhyllodocida (100%): PhyllodocidaNereididae (100%): NereididaePlatynereis (100%): PlatynereisHK01 (100%)
zotu.9 Root (100%): RootEukaryota (100%): EukaryotaAnnelida (100%): AnnelidaPolychaeta (100%): PolychaetaPhyllodocida (100%): PhyllodocidaNereididae (100%): NereididaePlatynereis (100%): PlatynereisHK01 (100%)
zotu.10 Root (100%): RootEukaryota (100%): EukaryotaArthropoda (100%): ArthropodaMalacostraca (100%): MalacostracaDecapoda (100%): DecapodaRhynchocinetidae (100%): RhynchocinetidaeRhynchocinetes (100%): Rhynchocinetesbrucei (58.3%)
The head output shows that the sequenceData/8-final/idtaxaTaxonomy.txt file is structured in two columns, with the first column specifying the ZOTU sequence name and the second column providing the taxonomic ID. Hence, a very similar structure to SINTAX, though the third column indicating the strand the match was made is missing, as well as the fourth column where SINTAX provided the taxonomic ID with the similarity threshold applied.
4.3 BLAST#
4.3.1 Online BLAST#
The third and final method we will explore is BLAST (Basic Local Alignment Search Tool), probably the most well known manner to assign a taxonomic ID to sequences. As the name suggests, BLAST assigns a taxonomic ID to a sequence through best hits from local alignments against a reference database.
The popularity of BLAST likely stems from its direct implementation into the NCBI online reference database repository. To check out the website, we will copy-paste the first 5 sequences in our ZOTU file (sequenceData/8-final/zotus.fasta) and perform a Nucleotide BLAST, as shown in the screenshot below.
Fig. 26 : A screenshot of the BLAST website#
Below are the top BLAST hit results for the first 5 sequences, followed by a screenshot of the website for the first ZOTU sequence (zotu.1).
Output
Results for: Description: Qcov: e-val: Pident:
zotu.1 (313 bp) Gammarus mucronatus 42% 4e-32 87.22%
zotu.2 (313 bp) Mitostoma chrysomelas 89% 2e-31 76.60%
zotu.3 (313 bp) Caridea sp. 99% 7e-90 86.58%
zotu.4 (313 bp) Caridea sp. 100% 2e-160 100.00%
zotu.5 (313 bp) Metaphire tosaensis 96% 9e-64 82.12%
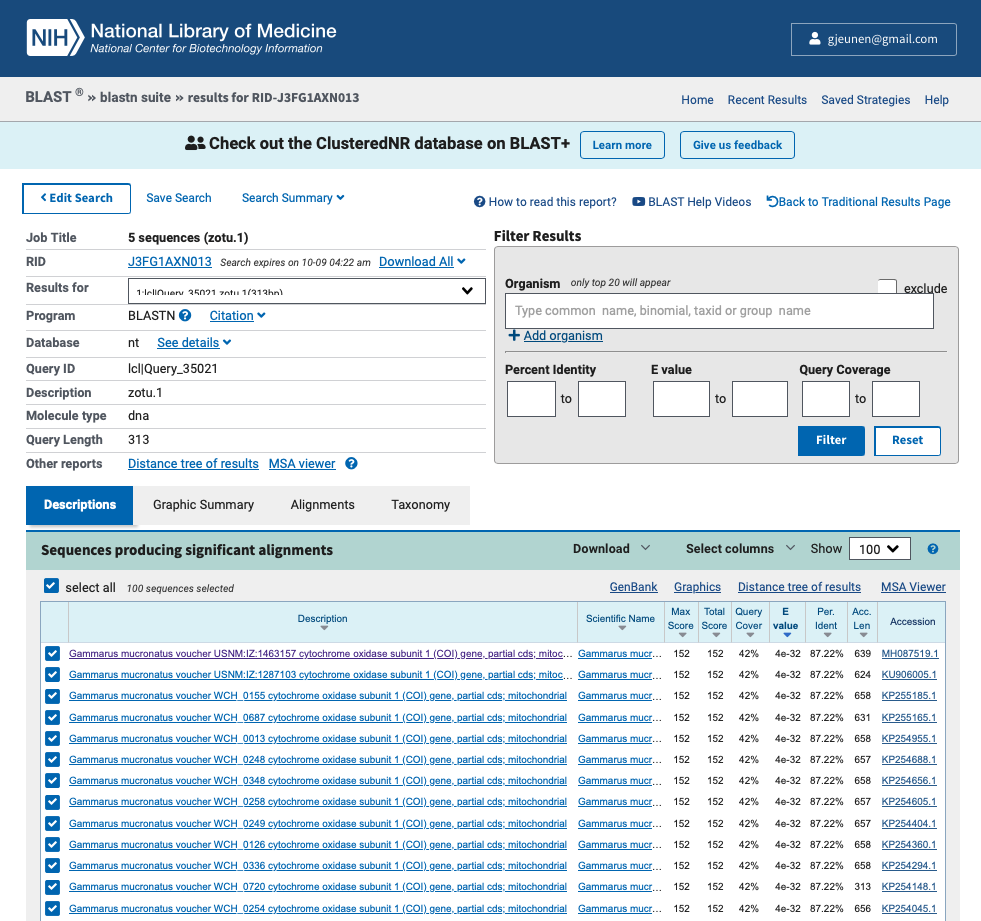
Fig. 27 : A screenshot of the results from the BLAST website for sequence ID zotu.1#
We will explore the results on the website more in depth during the workshop, but the most important numbers are provided in the output above, including the ZOTU sequence name (Results for:), the taxonomic ID the sequence best matches to (Description:), the percentage of how much the reference sequence provides coverage to the query sequence (Qcov:), the expect value is a parameter that describes the number of hits one can “expect” to see by chance when searching a database of a particular size and describes the random background noise (e-val:), and the percent identity value of how well the query and reference match over the covered region (Pident:).
4.3.2 BLAST+ remote#
While the website is extremely handy, it will be time-consuming if we have to manually check all 1,345 ZOTU sequences in our sequenceData/8-final/zotus.fasta file. Luckily, BLAST is also available via the Command Line Interface, called BLAST+. When we conduct BLAST via the command line (blastn), we can specify the parameter -remote, which will tell the program to access the NCBI servers and match your query sequences to the online NCBI database. To specify we want to search against the nucleotide database, we can provide the -db nt parameter. Our query sequences are provided through the -query parameter. We can also set the maximum number of target sequences we get back, the minimum percent identity threshold, and the minimum query coverage threshold through the -max_target_seqs, perc_identity, and -qcov_hsp_perc parameters. Finally, the output file (parameter: -out) can take on different formats, but the most common is a tab-delimited structure, which is invoked by -outfmt "6". Table C1 on this website provides an overview of all the different options to the BLAST+ search application.
To show you how you can run remote BLASTs in the Terminal, let’s extract the first five sequences from our sequenceData/8-final/zotus.fasta file and BLAST them remotely against the nucleotide NCBI database. To do all of this in the Terminal, rather than making a new document with the first five sequences, we can use the head command to extract the first five sequences and pipe the results to the blastn command.
Important
If you would like the scientific names provided in the output file, we first need to download and extract the taxonomic database from the NCBI website. We can do this using the following two commands
wget ftp://ftp.ncbi.nlm.nih.gov/blast/db/taxdb.tar.gz
tar -xvf taxdb.tar.gz
If you do not download the taxonomic database, the output file column sscinames in the code below will default to N/A.
head -n 25 sequenceData/8-final/zotus.fasta | blastn -db nt -query - -out sequenceData/8-final/BLASTremote.txt -max_target_seqs 10 -perc_identity 80 -qcov_hsp_perc 80 -remote -outfmt "6 qaccver saccver staxid sscinames length pident mismatch qcovs evalue bitscore qstart qend sstart send gapopen"
To display the output results of the blastn command in the Terminal window, we can use the cat command.
Important
We can use the cat command to display the contents of a file to the Terminal window. However, it is not recommended to do this for large files, as it will all be read into memory. For large files, it is best to use a command such as less.
cat sequenceData/8-final/BLASTremote.txt
Output
zotu.1 MH087519.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 500 632 0
zotu.1 KU906005.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 485 617 0
zotu.1 KP255185.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 519 651 0
zotu.1 KP255165.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 492 624 0
zotu.1 KP254955.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 519 651 0
zotu.1 KP254688.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 518 650 0
zotu.1 KP254656.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 519 651 0
zotu.1 KP254605.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 518 650 0
zotu.1 KP254404.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 518 650 0
zotu.1 KP254360.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 519 651 0
zotu.2 AB500136.1 322050 Neoseiulus womersleyi 167 82.036 29 53 9.72e-29 141 147 312 187 353 1
zotu.3 HM465067.1 874932 Caridea sp. LPdivOTU347 313 86.581 40 99 6.90e-90 344 2 313 294 605 2
zotu.4 HM466580.1 875001 Caridea sp. LPdivOTU96 313 100.000 0 100 1.74e-160 579 1 313 346 658 0
zotu.4 MZ560339.1 2858060 Hippolytidae sp. USNM 1466310 313 98.403 5 100 3.79e-152 551 1 313 346 658 0
zotu.4 MZ560299.1 2858059 Hippolytidae sp. USNM 1466309 313 98.403 5 100 3.79e-152 551 1 313 346 658 0
zotu.4 GQ260974.1 653025 Hippolytidae sp. LP-2009;Caridea sp. LPdivOTU96 313 98.403 5 100 3.79e-152 551 1 313 346 658 0
zotu.4 MZ560028.1 2858073 Hippolytidae sp. USNM 1466326 313 98.083 6 100 1.76e-150 545 1 313 346 658 0
zotu.4 MZ559959.1 2858071 Hippolytidae sp. USNM 1466317;Hippolytidae sp. USNM 1466318;Hippolytidae sp. USNM 1466319;Hippolytidae sp. USNM 1466320;Hippolytidae sp. USNM 1466321;Hippolytidae sp. USNM 1466322;Hippolytidae sp. USNM 1466323;Hippolytidae sp. USNM 1466324 313 98.083 6 100 1.76e-150 545 1 313 346 658 0
zotu.4 MZ559957.1 2858061 Hippolytidae sp. USNM 1466313 313 98.083 6 100 1.76e-150 545 1 313 346 658 0
zotu.4 MZ560029.1 2858074 Hippolytidae sp. USNM 1466328 311 98.071 6 99 2.28e-149 542 3 313 348 658 0
zotu.4 MZ560087.1 2858063 Hippolytidae sp. USNM 1466315 313 97.764 7 100 8.20e-149 540 1 313 346 658 0
zotu.4 MZ559958.1 2858064 Hippolytidae sp. USNM 1466316 313 97.764 7 100 8.20e-149 540 1 313 346 658 0
zotu.5 AB542683.1 506673 Metaphire tosaensis 302 82.119 52 96 9.25e-64 257 6 306 332 632 2
zotu.5 DQ224174.1 351234 Amynthas lini 302 81.788 53 96 4.30e-62 252 6 306 351 651 2
zotu.5 KM187649.1 1544308 Metabonellia haswelli 302 81.457 54 96 2.00e-60 246 6 306 351 651 2
zotu.5 HM219172.2 857735 Perionyx sp. 1 WL-2010 301 81.395 56 96 2.00e-60 246 6 306 350 650 0
zotu.5 MT472583.1 2736579 Perionyx sp. E1R2 301 81.063 57 96 9.32e-59 241 6 306 352 652 0
zotu.5 KT716821.1 1737888 Perionyx sp. DHS-2015 301 81.063 57 96 9.32e-59 241 6 306 362 662 0
zotu.5 LC703190.1 585715 Metaphire megascolidioides 302 81.126 55 96 9.32e-59 241 6 306 263 563 2
zotu.5 JF509718.1 6431 Urechis caupo 302 81.126 55 96 9.32e-59 241 6 306 350 650 2
zotu.5 AB542648.1 585715 Metaphire megascolidioides 302 81.126 55 96 9.32e-59 241 6 306 332 632 2
zotu.5 AB536863.1 585715 Metaphire megascolidioides 302 81.126 55 96 9.32e-59 241 6 306 332 632 2
The output shows similar results to the results we obtained from the website. However, now we immediately have the results in a tab-delimited text file that we can easily parse using a python script for example.
Important
It should be noted that if you ran BLAST via the website, you can use the BLAST+ command line software to download a similar table to your computer using the blast_formatter function. To accomplish this, you would need to provide the RID tag with the -rid parameter. Below is a screenshot on where you can locate this RID tag on the website and the code to download the BLAST hits of the 5 sequences we checked on the website. Online BLAST results typically stay available through the RID tag for 24 - 48h. While you can set the maximum number of sequences per entry to be downloaded -max_target_seqs, this method does not allow the user to set a percent identity or query coverage threshold.
Fig. 28 : A screenshot of where to locate the RID tag on the NCBI website to download online BLAST results#
blast_formatter -rid J3FG1AXN013 -out sequenceData/8-final/blastonline.txt -max_target_seqs 10 -outfmt "6 qaccver saccver staxid sscinames length pident mismatch qcovs evalue bitscore qstart qend sstart send gapopen"
cat sequenceData/8-final/BLASTonline.txt
Output
zotu.1 MH087519.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 500 632 0
zotu.1 KU906005.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 485 617 0
zotu.1 KP255185.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 519 651 0
zotu.1 KP255165.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 492 624 0
zotu.1 KP254955.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 519 651 0
zotu.1 KP254688.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 518 650 0
zotu.1 KP254656.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 519 651 0
zotu.1 KP254605.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 518 650 0
zotu.1 KP254404.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 518 650 0
zotu.1 KP254360.1 315708 Gammarus mucronatus 133 87.218 17 42 4.49e-32 152 174 306 519 651 0
zotu.2 KY268691.1 673248 Mitostoma chrysomelas 282 76.596 60 89 1.62e-31 150 29 307 374 652 5
zotu.2 KY268314.1 673248 Mitostoma chrysomelas 282 76.241 61 89 2.09e-30 147 29 307 374 652 5
zotu.2 OK072767.1 2899957 Paraleptamphopidae sp. NZGDW0096 261 77.011 56 82 2.09e-30 147 50 307 376 635 4
zotu.2 OK072738.1 2899963 Paraleptamphopidae sp. NZGDW0110 261 77.011 56 82 2.09e-30 147 50 307 343 602 4
zotu.2 JF444969.1 38668 Onychomys leucogaster 273 76.923 51 85 7.51e-30 145 29 295 373 639 10
zotu.2 OK072800.1 2899962 Paraleptamphopidae sp. NZGDW0107 261 76.628 57 82 2.70e-29 143 50 307 343 602 4
zotu.2 AB500136.1 322050 Neoseiulus womersleyi 167 82.036 29 53 9.72e-29 141 147 312 187 353 1
zotu.2 KY269411.1 673248 Mitostoma chrysomelas 282 75.887 62 89 3.50e-28 139 29 307 374 652 5
zotu.2 KY269355.1 673248 Mitostoma chrysomelas 282 75.887 62 89 3.50e-28 139 29 307 374 652 5
zotu.2 KY268900.1 673248 Mitostoma chrysomelas 282 75.887 62 89 3.50e-28 139 29 307 374 652 5
zotu.3 HM465067.1 874932 Caridea sp. LPdivOTU347 313 86.581 40 99 6.90e-90 344 2 313 294 605 2
zotu.3 MG093570.1 2362403 Mycetophila sp. BIOUG21226-D02 302 79.470 58 95 7.30e-50 211 13 312 357 656 2
zotu.3 KR651281.1 1881779 Mycetophila sp. BOLD-2016 302 79.139 59 95 3.40e-48 206 13 312 358 657 2
zotu.3 KM960518.1 1750023 Mycetophilidae sp. BOLD:ACG6335 302 78.808 60 95 1.58e-46 200 13 312 358 657 2
zotu.3 MN868958.1 2690821 Rhamphomyia dudai 300 78.333 63 95 2.65e-44 193 14 312 359 657 2
zotu.3 MT826940.1 149068 Nematoscelis megalops 299 78.261 65 95 2.65e-44 193 14 312 373 671 0
zotu.3 KM978363.1 1302337 Orchesellides sinensis 300 78.000 66 95 3.42e-43 189 14 313 359 658 0
zotu.3 KR039462.1 30126 Graminella mohri 304 77.961 63 96 1.23e-42 187 11 312 362 663 2
zotu.3 KM023138.1 82746 Orius niger 305 78.033 61 96 1.23e-42 187 11 312 356 657 5
zotu.3 KM022126.1 82746 Orius niger 305 78.033 61 96 1.23e-42 187 11 312 101 402 5
zotu.4 HM466580.1 875001 Caridea sp. LPdivOTU96 313 100.000 0 100 1.74e-160 579 1 313 346 658 0
zotu.4 MZ560339.1 2858060 Hippolytidae sp. USNM 1466310 313 98.403 5 100 3.79e-152 551 1 313 346 658 0
zotu.4 MZ560299.1 2858059 Hippolytidae sp. USNM 1466309 313 98.403 5 100 3.79e-152 551 1 313 346 658 0
zotu.4 GQ260974.1 653025 Hippolytidae sp. LP-2009;Caridea sp. LPdivOTU96 313 98.403 5 100 3.79e-152 551 1 313 346 658 0
zotu.4 MZ560028.1 2858073 Hippolytidae sp. USNM 1466326 313 98.083 6 100 1.76e-150 545 1 313 346 658 0
zotu.4 MZ559959.1 2858071 Hippolytidae sp. USNM 1466317;Hippolytidae sp. USNM 1466318;Hippolytidae sp. USNM 1466319;Hippolytidae sp. USNM 1466320;Hippolytidae sp. USNM 1466321;Hippolytidae sp. USNM 1466322;Hippolytidae sp. USNM 1466323;Hippolytidae sp. USNM 1466324 313 98.083 6 100 1.76e-150 545 1 313 346 658 0
zotu.4 MZ559957.1 2858061 Hippolytidae sp. USNM 1466313 313 98.083 6 100 1.76e-150 545 1 313 346 658 0
zotu.4 MZ560029.1 2858074 Hippolytidae sp. USNM 1466328 311 98.071 6 99 2.28e-149 542 3 313 348 658 0
zotu.4 MZ560087.1 2858063 Hippolytidae sp. USNM 1466315 313 97.764 7 100 8.20e-149 540 1 313 346 658 0
zotu.4 MZ559958.1 2858064 Hippolytidae sp. USNM 1466316 313 97.764 7 100 8.20e-149 540 1 313 346 658 0
zotu.5 AB542683.1 506673 Metaphire tosaensis 302 82.119 52 96 9.25e-64 257 6 306 332 632 2
zotu.5 DQ224174.1 351234 Amynthas lini 302 81.788 53 96 4.30e-62 252 6 306 351 651 2
zotu.5 KM187649.1 1544308 Metabonellia haswelli 302 81.457 54 96 2.00e-60 246 6 306 351 651 2
zotu.5 HM219172.2 857735 Perionyx sp. 1 WL-2010 301 81.395 56 96 2.00e-60 246 6 306 350 650 0
zotu.5 MT472583.1 2736579 Perionyx sp. E1R2 301 81.063 57 96 9.32e-59 241 6 306 352 652 0
zotu.5 KT716821.1 1737888 Perionyx sp. DHS-2015 301 81.063 57 96 9.32e-59 241 6 306 362 662 0
zotu.5 LC703190.1 585715 Metaphire megascolidioides 302 81.126 55 96 9.32e-59 241 6 306 263 563 2
zotu.5 JF509718.1 6431 Urechis caupo 302 81.126 55 96 9.32e-59 241 6 306 350 650 2
zotu.5 AB542648.1 585715 Metaphire megascolidioides 302 81.126 55 96 9.32e-59 241 6 306 332 632 2
zotu.5 AB536863.1 585715 Metaphire megascolidioides 302 81.126 55 96 9.32e-59 241 6 306 332 632 2
4.3.3 BLAST+ local#
While running BLAST+ remotely is very handy, it is frowned upon to run remote BLAST searches for thousands of sequences and should be restricted to a couple of sequences at a time. The reason for this is that we don’t want to abuse our priviledge to take up too much of the traffic on the NCBI servers. One way to circumvent the issue is to set up a script that iterates over our 1,345 ZOTU sequences to run remote BLAST searches separately. Similarly, posting a file with thousands of sequences for an online BLAST will encounter time-out errors.
Hence, when dealing with large numbers of sequences, it is best to run BLAST+ on a local database. If you are working on a supercomputer, they might have a locally installed and up-to-date version of the full NCBI database available to you. This option, however, is not available to everyone. Besides BLASTing against the full NCBI database (which is a couple of TB in size), we could also use the database we created with CRABS to run BLAST locally. Since running BLAST on a curated reference database you have built yourself is more accessible to everyone than a local version of the full NCBI database on a supercomputer, we will use the curated database option for this tutorial.
The first step before running BLAST, is to reformat the initial database, similarly to what we had to do before we could run the IDTAXA machine learning algorithm. To generate a local BLAST database, we first need to remove the SINTAX-specific formatting of our database. We can do this using the following python script, which will remove all the header information and only keep the accession numbers as sequence headers. This python script takes in two user arguments, including the input file and an output file name for the BLAST database.
nano sequenceData/1-scripts/blastInitFormat.py
#! /usr/bin/env python3
import sys
inputFile = sys.argv[1]
outputFile = sys.argv[2]
newDict = {}
with open(inputFile, 'r') as infile:
for line in infile:
if line.startswith('>'):
seqID = line.split(';')[0] + '\n'
if 'CRABS_' in seqID:
seqID = seqID.split(':')[0] + '\n'
else:
newDict[seqID] = line
with open(outputFile, 'w') as outfile:
for k, v in newDict.items():
_ = outfile.write(k)
_ = outfile.write(v)
Press ctrl + x to exit out of the editor, followed by y and return.
chmod +x sequenceData/1-scripts/blastInitFormat.py
./sequenceData/1-scripts/blastInitFormat.py sequenceData/7-refdb/newDBcrabsCOIsintax.fasta sequenceData/7-refdb/blast.fasta
Once we have generated a fasta file with the correct format, we can create a BLAST database using the makeblastdb command in the BLAST+ suite.
makeblastdb -in sequenceData/7-refdb/blast.fasta -dbtype nucl -parse_seqids -out sequenceData/7-refdb/blastDBCOI
Output
Building a new DB, current time: 10/10/2023 10:26:06
New DB name: /Users/gjeunen/Documents/work/lectures/HKU2023/tutorial/sequenceData/7-refdb/blastDBCOI
New DB title: sequenceData/7-refdb/blast.fasta
Sequence type: Nucleotide
Deleted existing Nucleotide BLAST database named /Users/gjeunen/Documents/work/lectures/HKU2023/tutorial/sequenceData/7-refdb/blastDBCOI
Keep MBits: T
Maximum file size: 1000000000B
Adding sequences from FASTA; added 208189 sequences in 3.29866 seconds.
We can now use blastn locally to assign a taxonomic ID to each ZOTU sequence using the BLAST algorithm.
blastn -query sequenceData/8-final/zotus.fasta -db sequenceData/7-refdb/blastDBCOI -max_target_seqs 10 -perc_identity 50 -qcov_hsp_perc 50 -outfmt "6 qaccver saccver staxid sscinames length pident mismatch qcovs evalue bitscore qstart qend sstart send gapopen" -out sequenceData/8-final/blastTaxonomy.txt
By using the head command, we can inspect the first couple of lines of the output document.
head -n 20 sequenceData/8-final/blastTaxonomy.txt
Output
zotu.1 KT708260 N/A N/A 297 75.421 73 95 4.53e-34 145 11 307 356 652 0
zotu.1 KT707216 N/A N/A 297 75.421 73 95 4.53e-34 145 11 307 356 652 0
zotu.1 MF744717 N/A N/A 252 76.190 58 80 3.53e-30 132 57 307 402 652 2
zotu.2 KY268691 N/A N/A 282 76.596 60 89 9.74e-36 150 29 307 374 652 5
zotu.2 OK072767 N/A N/A 261 77.011 56 82 1.26e-34 147 50 307 376 635 4
zotu.2 OK072738 N/A N/A 261 77.011 56 82 1.26e-34 147 50 307 343 602 4
zotu.2 KY268314 N/A N/A 282 76.241 61 89 1.26e-34 147 29 307 374 652 5
zotu.2 OK072800 N/A N/A 261 76.628 57 82 1.63e-33 143 50 307 343 602 4
zotu.2 KY269355 N/A N/A 282 75.887 62 89 2.11e-32 139 29 307 374 652 5
zotu.2 KY268900 N/A N/A 282 75.887 62 89 2.11e-32 139 29 307 374 652 5
zotu.2 OK072832 N/A N/A 261 76.245 58 82 2.73e-31 135 50 307 355 614 4
zotu.2 OK072819 N/A N/A 261 76.245 58 82 2.73e-31 135 50 307 348 607 4
zotu.2 OK072786 N/A N/A 261 76.245 58 82 2.73e-31 135 50 307 376 635 4
zotu.3 BHKG-0179 N/A N/A 313 99.681 1 100 4.87e-163 573 1 313 346 658 0
zotu.3 HM465067 N/A N/A 313 86.581 40 99 4.16e-94 344 2 313 294 605 2
zotu.3 MT826940 N/A N/A 299 78.261 65 96 1.59e-48 193 14 312 373 671 0
zotu.3 MT826939 N/A N/A 299 77.926 66 96 7.42e-47 187 14 312 373 671 0
zotu.3 MT826938 N/A N/A 299 77.926 66 96 7.42e-47 187 14 312 373 671 0
zotu.3 MT826937 N/A N/A 293 78.157 64 94 7.42e-47 187 14 306 373 665 0
zotu.3 MT826936 N/A N/A 299 77.926 66 96 7.42e-47 187 14 312 373 671 0
From the head output, we can see that the local BLAST output file follows the same structure as the remote BLAST output file. However, note the missing values for the taxonomic ID and species names! We have excluded this information when we generated the database, as errors occur when using custom barcodes for which no accession numbers exist. No worries about this though, as we will generate the taxonomic ID, species name, and taxonomic lineage for each BLAST hit when parsing the output file in section 5.3 BLAST formatting.
Important
For people who’s reference database solely exists out of NCBI sequences with valid accession numbers, you can also generate a reference database that includes the taxonomic ID and species name information. When the reference database is built in such a manner the blastn output will contain the taxonomic ID and species name values, rather than defaulting to N/A. Provided below is the code to build such a database.
If we would like to add the taxonomic ID and scientific names in the blastn output file, as in the blastn -remote option, we will need to run some additional lines of code before using the makeblastdb command and ensure only valid accession numbers are in the reference database and incorporated in the nucl_gb.accession2taxid (which we downloaded when building a reference database with CRABS in section 2. Reference databases).
Note
If you would like to have the taxonomic ID and scientific name outputs, but your reference database includes invalid accession number, i.e., non-NCBI sequences, you can add your sequence ID’s to the nucl_gb.accession2taxid file, which is a simple tab-delimited text file that matches accession numbers to NCBI taxIDs. This will need to be done manually, though.
First, we need to generate an accession number to taxonomic ID mapping file. This can be done by removing the second and fourth column from the nucl_gb.accession2taxid file we downloaded when building a reference database with CRABS in section 2. Reference databases.
tail -n +2 sequenceData/7-refdb/nucl_gb.accession2taxid | awk '{print $1, $3}' > sequenceData/7-refdb/taxIDmap.txt
Second, if you haven’t downloaded the taxonomic NCBI database during section 4.3.2 BLAST+ remote, you need to run the following two commands (this can be skipped if you already downloaded these files).
wget ftp://ftp.ncbi.nlm.nih.gov/blast/db/taxdb.tar.gz
tar -xvf taxdb.tar.gz
Third, we can run an altered version of the makeblastdb command, where we specify the sequenceData/7-refdb/taxIDmap.txt file using the -taxid_map parameter.
makeblastdb -in sequenceData/7-refdb/blast.fasta -dbtype nucl -parse_seqids -out sequenceData/7-refdb/blastDBCOI -taxid_map sequenceData/7-refdb/taxIDmap.txt
Once this command is executed, you can run the blastn command with an output that specifies the taxonomic ID and species name values for each BLAST hit.
5. Output formatting#
At this stage, we have explored three taxonomy assignment algorithms and created multiple files containing the taxonomic ID for each ZOTU sequence, including sequenceData/8-final/sintaxTaxonomy.txt for the SINTAX classifier, sequenceData/8-final/idtaxaTaxonomy.txt for the IDTAXA classifier, and sequenceData/8-final/blastTaxonomy.txt for the BLAST classifier. Each of these files has a unique, but similar structure. For the data exploration and statistical analyses in R, it would be easiest if all have the same structure, so they can be read in using the same code. Therefore, let’s reformat all files to a unique format, so they can be easily compared and read into R. We will be using some simple python parsing scripts to accomplish this goal.
5.1 SINTAX formatting#
The SINTAX output is a tab-delimited text file, where the first column contains the ZOTU identifiers. These identifiers are not ordered based on number, though all ZOTU numbers are present in the document, even when no taxonomic ID could be assigned. The second column contains the taxonomic ID information, including lineage and confidence values. The third column denotes the DNA strand the assignment was made. The fourth column and last column contains the taxonomic lineage up to the point where the confidence value is larger than the cut off value, 70% in our case. We can use the python script below to parse the SINTAX document. This python script takes five user arguments, including the SINTAX input file, an output file name for the new lineage document, an output file name for the new score document, the number of ZOTUs, and the similarity threshold value.
nano sequenceData/1-scripts/sintaxOutputFormat.py
#! /usr/bin/env python3
import sys
import collections
sintaxInputFile = sys.argv[1]
sintaxLineageOutputFile = sys.argv[2]
sintaxScoreOutputFile = sys.argv[3]
zotuNumber = int(sys.argv[4])
cutOffValue = float(sys.argv[5])
headerRanks = ['kingdom', 'phylum', 'class', 'order', 'family', 'genus', 'species']
idLineageDict = collections.defaultdict(dict)
idScoreDict = collections.defaultdict(dict)
with open(sintaxInputFile, 'r') as inputFile:
for line in inputFile:
seqID = line.split('\t')[0]
lineage = line.split('\t')[1]
lineageItems = lineage.split(',')
if len(lineageItems) == 1:
for item in headerRanks:
idLineageDict[seqID][item] = 'NA'
idScoreDict[seqID][item] = 'NA'
else:
for item in range(len(lineageItems)):
rankID = lineageItems[item].split(':')[1].split('(')[0]
scoreID = lineageItems[item].split('(')[-1].split(')')[0]
if float(scoreID) >= cutOffValue:
idLineageDict[seqID][headerRanks[item]] = rankID
idScoreDict[seqID][headerRanks[item]] = scoreID
else:
idLineageDict[seqID][headerRanks[item]] = 'NA'
idScoreDict[seqID][headerRanks[item]] = scoreID
with open(sintaxLineageOutputFile, 'w') as outfile:
_ = outfile.write('ID' + '\t' + '\t'.join(headerRanks) + '\n')
for i in range(zotuNumber):
_ = outfile.write(f'zotu.{i+1}')
for k, v in idLineageDict[f'zotu.{i+1}'].items():
_ = outfile.write('\t' + v)
_ = outfile.write('\n')
with open(sintaxScoreOutputFile, 'w') as outfile:
_ = outfile.write('ID' + '\t' + '\t'.join(headerRanks) + '\n')
for i in range(zotuNumber):
_ = outfile.write(f'zotu.{i+1}')
for k, v in idScoreDict[f'zotu.{i+1}'].items():
_ = outfile.write('\t' + v)
_ = outfile.write('\n')
Press ctrl + x to exit out of the editor, followed by y and return.
chmod +x sequenceData/1-scripts/sintaxOutputFormat.py
./sequenceData/1-scripts/sintaxOutputFormat.py sequenceData/8-final/sintaxTaxonomy.txt sequenceData/8-final/sintaxLineage.txt sequenceData/8-final/sintaxScore.txt 1345 0.7
Opening both documents shows that we have created two tab-delimited text files, with sequenceData/8-final/sintaxLineage.txt containing the lineage information of the taxonomic ID and sequenceData/8-final/sintaxScore.txt containing the confidence score of the taxonomic ID for each level.
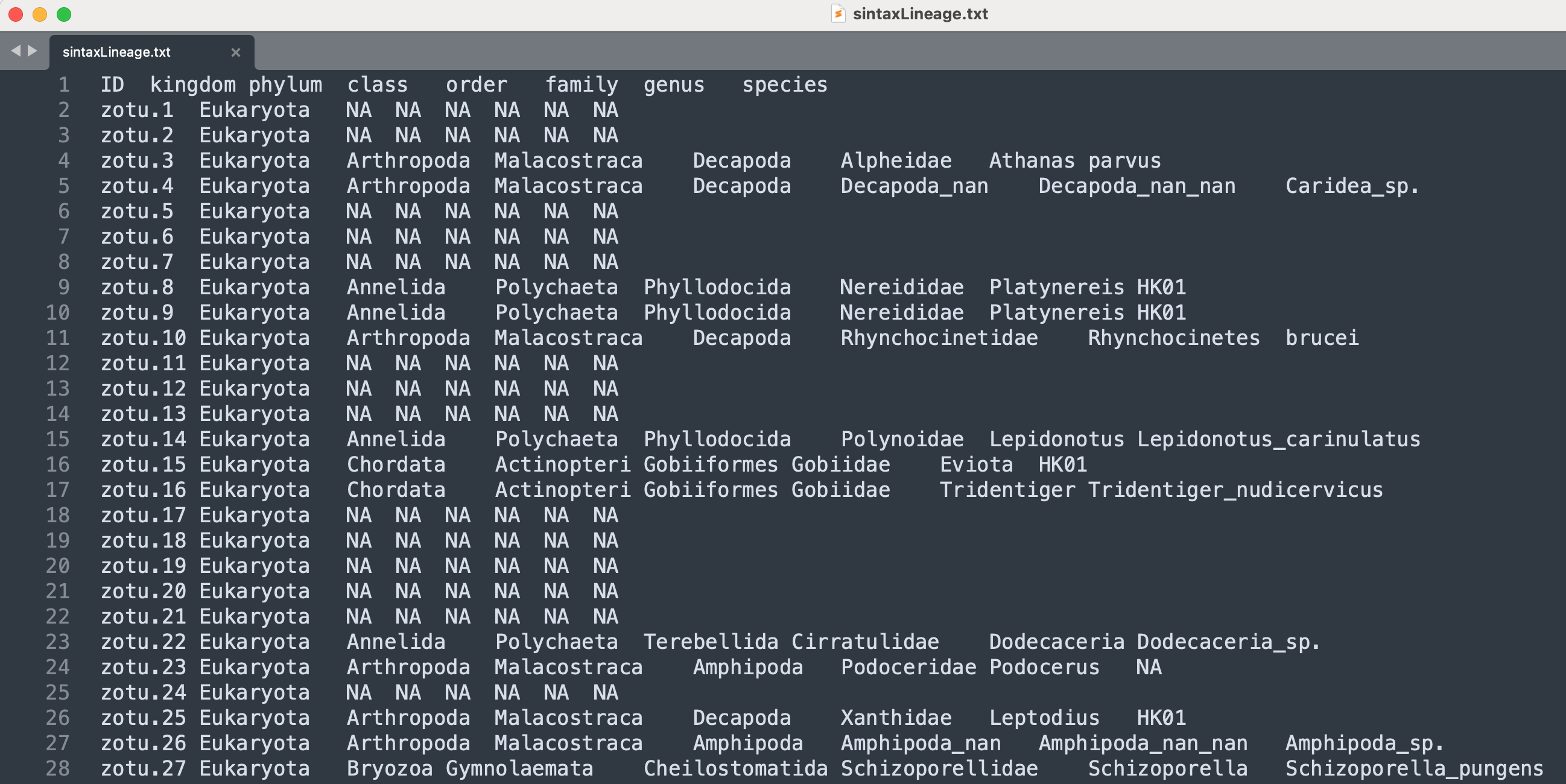
Fig. 29 : A screenshot of the SINTAX results after parsing the output document.#
Note
Note that we set a manual confidence threshold of 70% and have set the taxonomic ID to NA if this value wasn’t reached. Changing the value in the python script allows you to change the similarity threshold after SINTAX classification to see how different thresholds impact the final taxonomic ID.
5.2 IDTAXA formatting#
The IDTAXA output is very similarly structured as the SINTAX output, except that the first column is ordered on ZOTU identifiers, the second column is slightly differently formatted but contains the same information, and the third and fourth columns are missing. We can use the python script below to format the IDTAXA file, which takes three user arguments, including the input file, an output file name for the new lineage document, and an output file name for the new score document. As the ZOTU sequences are ordered and ZOTU sequences are included even when no taxonomic ID could be achieved, we do not need to provide the total number of sequences in this python script.
Note
This python document also reverts the “hack” used with homonym names.
nano sequenceData/1-scripts/idtaxaOutputFormat.py
#! /usr/bin/env python3
import sys
import collections
def removeBeforeSecondUppercase(input_string):
uppercase_count = 0
result = ''
count = 0
for char in input_string:
count += 1
if char.isupper():
uppercase_count += 1
if uppercase_count == 2:
result = input_string[count -1:]
break
return result
idTaxaInputFile = sys.argv[1]
idTaxaLineageOutputFile = sys.argv[2]
idTaxaScoreOutputFile = sys.argv[3]
headerRanks = ['root', 'kingdom', 'phylum', 'class', 'order', 'family', 'genus', 'species']
idLineageDict = collections.defaultdict(dict)
idScoreDict = collections.defaultdict(dict)
with open(idTaxaInputFile, 'r') as inputFile:
for line in inputFile:
line = line.rstrip('\n')
seqID = line.split('\t')[0]
lineage = line.split('\t')[1]
lineageItems = lineage.split(': ')
for item in range(len(headerRanks)):
try:
if 'unclassified' in lineageItems[item]:
idLineageDict[seqID][headerRanks[item]] = 'NA'
idScoreDict[seqID][headerRanks[item]] = 'NA'
else:
lineageString = lineageItems[item].split(' ')[0]
if lineageString != 'Root':
lineageString = removeBeforeSecondUppercase(lineageString)
scoreString = lineageItems[item].split('(')[1].rstrip(')')
idScoreDict[seqID][headerRanks[item]] = scoreString
idLineageDict[seqID][headerRanks[item]] = lineageString
except IndexError:
idLineageDict[seqID][headerRanks[item]] = 'NA'
idScoreDict[seqID][headerRanks[item]] = 'NA'
with open(idTaxaLineageOutputFile, 'w') as outfile:
_ = outfile.write('ID' + '\t' + '\t'.join(headerRanks) + '\n')
for i in idLineageDict:
_ = outfile.write(i)
for j, g in idLineageDict[i].items():
_ = outfile.write('\t' + g)
_ = outfile.write('\n')
with open(idTaxaScoreOutputFile, 'w') as outfile:
_ = outfile.write('ID' + '\t' + '\t'.join(headerRanks) + '\n')
for i in idScoreDict:
_ = outfile.write(i)
for j, g in idScoreDict[i].items():
_ = outfile.write('\t' + g)
_ = outfile.write('\n')
Press ctrl + x to exit out of the editor, followed by y and return.
chmod +x sequenceData/1-scripts/idtaxaOutputFormat.py
./sequenceData/1-scripts/idtaxaOutputFormat.py sequenceData/8-final/idtaxaTaxonomy.txt sequenceData/8-final/idtaxaLineage.txt sequenceData/8-final/idtaxaScore.txt
Similarly to the SINTAX parsing, opening both documents shows that we have created two tab-delimited text files, with sequenceData/8-final/idtaxaLineage.txt containing the lineage information of the taxonomic ID and sequenceData/8-final/idtaxaScore.txt containing the confidence score of the taxonomic ID for each level.
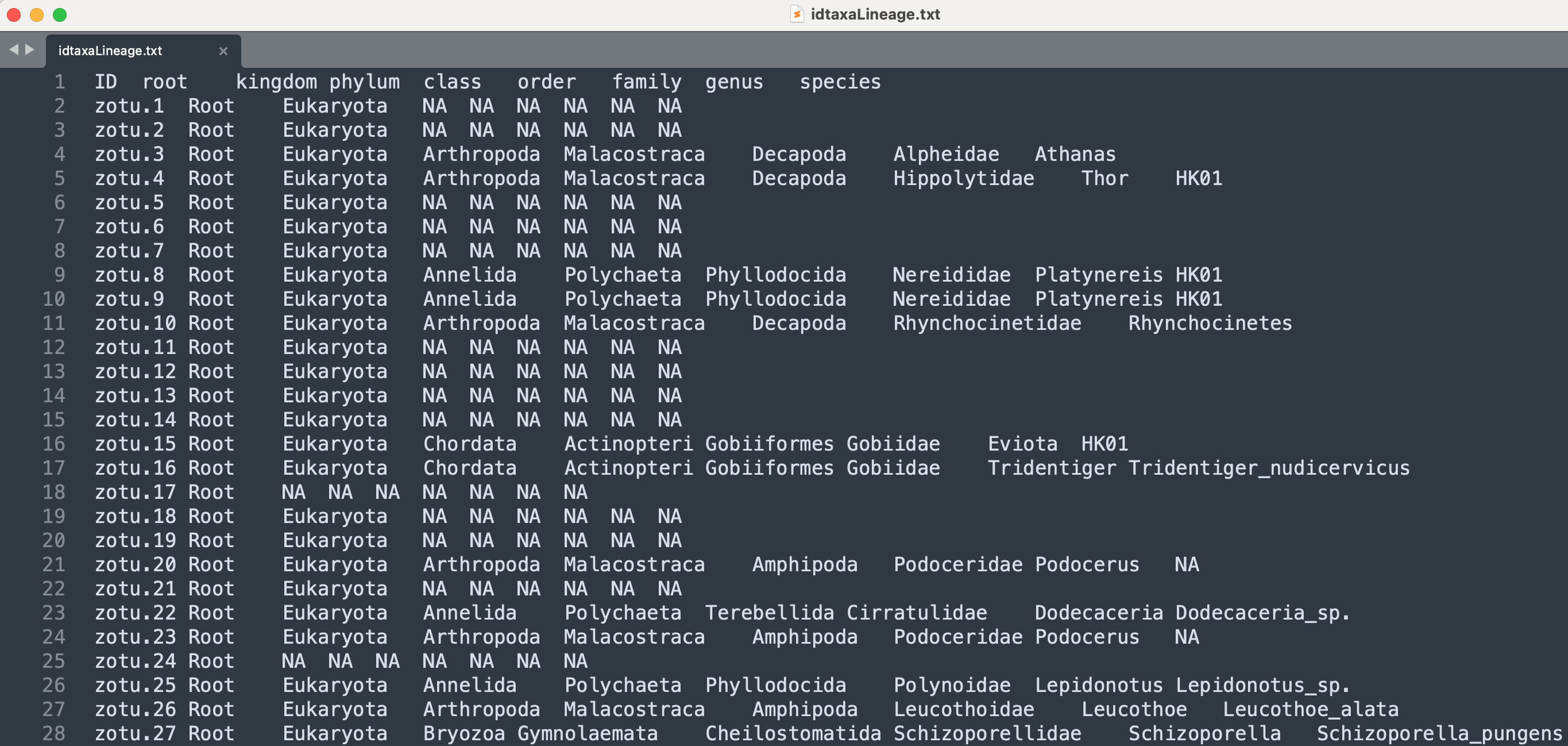
Fig. 30 : A screenshot of the IDTAXA results after parsing the output document.#
5.3 BLAST formatting#
The BLAST output is a tab-delimited text file, whereby the first column is an ordered ZOTU identifier list. However, ZOTUs for which no match was found are missing. The second column provides the accession number for the best match. The fourth column should contain the species name, but is defaulted to N/A as we used in house barcodes (see section 4.3.3 BLAST+ local) and the following columns report the statistics for the BLAST match. We can use the python script below to parse the document. It takes four user arguments, including the BLAST input file, the file name for the output document, the number of ZOTUs, and the original sintax-formatted reference database (sequenceData/7-refdb/newDBcrabsCOIsintax.fasta) to retrieve the species name and taxonomic lineage for each accession number.
Warning
The following python code only takes into account the top BLAST hit to provide an example of how to parse BLAST documents. For your own research project, I do not recommend relying solely on top BLAST hits, but rather determine the MRCA (Most Recent Common Ancestor) from the BLAST output first. Generating an MRCA is outside the scope of this tutorial. For more information on how to generate MRCA taxonomic IDs from BLAST output files, please have a look at this GitHub repo on MRCA calculation I have written a while ago. It contains a full working software program to calculate MRCAs from BLAST output files.
nano sequenceData/1-scripts/blastOutputFormat.py
#! /usr/bin/env python3
import sys
import subprocess as sp
import collections
blastInputFile = sys.argv[1]
BLASTOutputFile = sys.argv[2]
numberOfZotus = int(sys.argv[3])
refDB = sys.argv[4]
zotuIDblastDict = collections.defaultdict(dict)
zotuDict = {}
accessionDict = {}
with open(refDB, 'r') as refFile:
for line in refFile:
if line.startswith('>'):
accessionNumber = line.split(';')[0].lstrip('>')
if accessionNumber.startswith('CRABS_'):
accessionNumber = accessionNumber.split(':')[0]
tax = line.split('=')[-1].rstrip('\n').split(',')
for i in range(len(tax)):
tax[i] = tax[i].split(':', 2)[-1]
taxLineage = '\t'.join(tax)
accessionDict[accessionNumber] = taxLineage
with open(blastInputFile, 'r') as infile:
for line in infile:
zotuID = line.split('\t')[0]
if zotuID in zotuDict:
continue
zotuDict[zotuID] = 1
pident = line.split('\t')[5]
qcov = line.split('\t')[7]
acc = line.split('\t')[1]
zotuIDblastDict[zotuID]['pident'] = pident
zotuIDblastDict[zotuID]['qcov'] = qcov
zotuIDblastDict[zotuID]['accession'] = acc
with open(BLASTOutputFile, 'w') as outfile:
_ = outfile.write('ID' + '\t' 'kingdom' + '\t' 'phylum' + '\t' 'class' + '\t' 'order' + '\t' 'family' + '\t' 'genus' + '\t' 'species' + '\t' + 'pident' + '\t' + 'qcov' + '\n')
for i in range(numberOfZotus):
if len(zotuIDblastDict[f'zotu.{i+1}']) == 0:
_ = outfile.write(f'zotu.{i+1}' + '\tNA\tNA\tNA\tNA\tNA\tNA\tNA\tNA\tNA\n')
else:
_ = outfile.write(f'zotu.{i+1}')
accessionN = zotuIDblastDict[f'zotu.{i+1}']['accession']
pidentN = zotuIDblastDict[f'zotu.{i+1}']['pident']
qcovN = zotuIDblastDict[f'zotu.{i+1}']['qcov']
_ = outfile.write(f'\t{accessionDict[accessionN]}\t{pidentN}\t{qcovN}\n')
Press ctrl + x to exit out of the editor, followed by y and return.
chmod +x sequenceData/1-scripts/blastOutputFormat.py
./sequenceData/1-scripts/blastOutputFormat.py sequenceData/8-final/blastTaxonomy.txt sequenceData/8-final/blastLineage.txt 1345 sequenceData/7-refdb/newDBcrabsCOIsintax.fasta
Opening the parsed BLAST document shows that we have created a tab-delimited text file, containing the ZOTU sequence ID in the first column, species ID in the second column, percent identity value in the third column, and query coverage value in the fourth column. Note the difference in sequences that have a BLAST hit compared to the SINTAX and IDTAXA classifiers, even though we used the same reference database! This is not due to BLAST being a better classifier, rather than our low thresholds for allowing a BLAST hit to be accepted. Increasing the percent identity and query coverage value to SINTAX or IDTAXA standard will result in a similar drop out of sequences without a taxonomic ID.
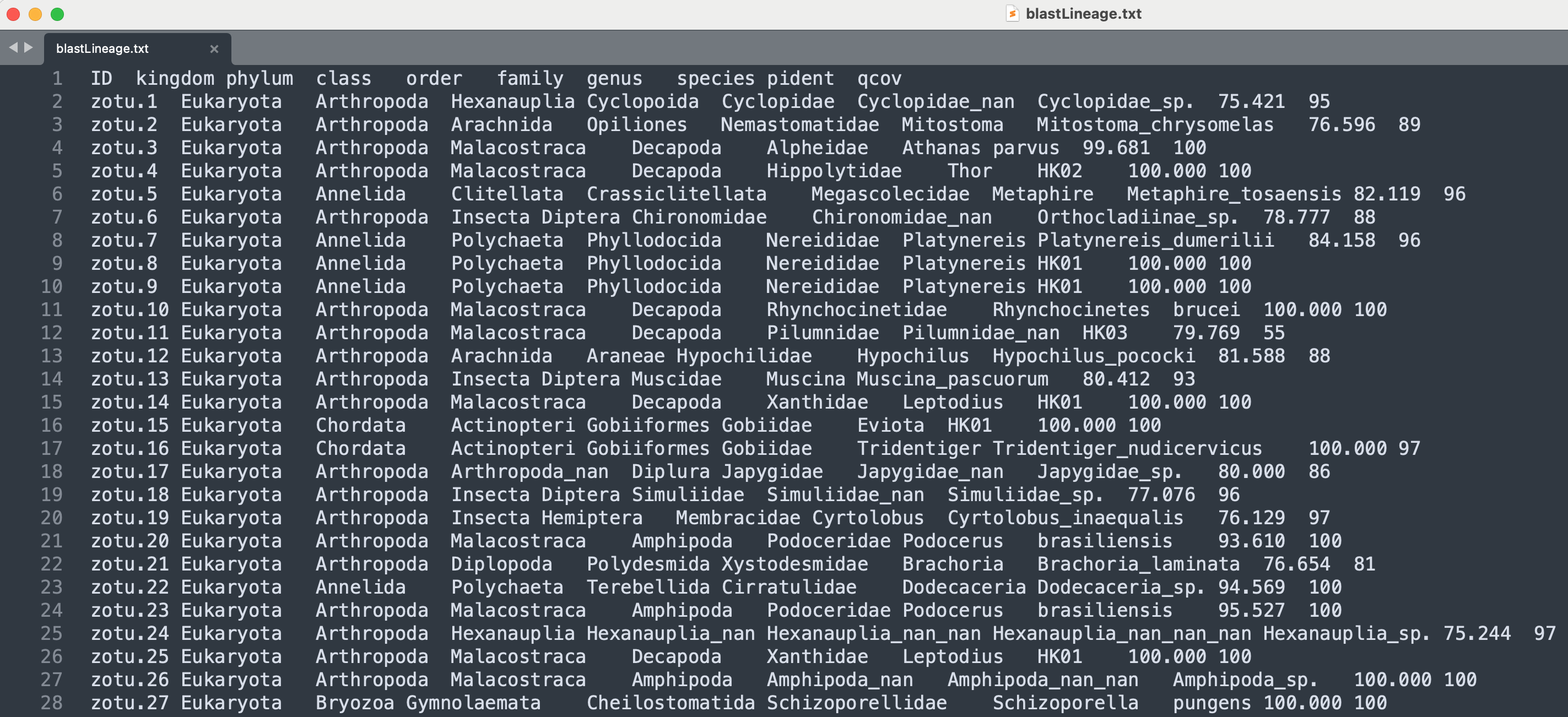
Fig. 31 : A screenshot of the BLAST results after parsing the output document.#
That’s it for today, see you tomorrow when we will explore the data and conduct statistical analyses to determine differences between samples in our tutorial data set!