Data pre-processing#
1. Introduction#
Before exploring the data and conducting the statistical analysis, we need to perform some pre-processing steps on our frequency table to clean up the data. Within the eDNA metabarcoding research community, there is no standardised manner of pre-processing data yet. However, steps that are frequently included are (list is not in order of importance):
abundance filtering of specific detections to eliminate the risk of barcode hopping signals interfering with the analysis
usually setting singleton detections to 0;
sometimes an abundance threshold is used, but this requires a positive control sample to determine the appropriate threshold.
filtering of ZOTUs which have a positive detection in the negative controls
either by complete removal of the ZOTU;
or based on an abundance threshold obtained from the read count in the negative controls;
or through a statistical test.
removal of low-confidence taxonomy assignments (if analysis is taxonomy-dependent)
discard samples with low read count.
removal of artificial ZOTUs
taxon-independent merging through the LULU algorithm;
taxon-dependent merging.
transforming raw read count
to relative abundance;
presence-absence;
or other transformations.
Important
From this point onwards, most code will be run in an R environment, such as RStudio, unless specified otherwise. Therefore, I recommend to open a new R-script, copy-paste the provided R code below into it, and save this script as sequenceData/1-scripts/dataPreProcessing.R.
Danger
Please note that my preferred coding language is Python. Due to student’s preference for R, however, the pre-processing and statistical analyses for this tutorial will be conducted in RStudio. Be mindful that due to my limited experience with R, some code might not be as optimal as it can be. All code should execute without bugging out though.
Remember that all code we will run in R today, you can just as easily run in Python (in case someone is interested)!
To get us started with the R environment, let’s open up a new R-script, set up the working directory and load the packages we will be using for the analysis.
#########################
# PREPARE R ENVIRONMENT #
#########################
library(Biostrings)
library(dplyr)
library(decontam)
library(phyloseq)
library(ggplot2)
library(lulu)
library(microbiome)
library(scales)
library(vegan)
library(ampvis2)
library(tidyverse)
# set working directory
setwd('/Users/gjeunen/Documents/work/lectures/HKU2023/tutorial/sequenceData/8-final')
2. Read Data into R#
First things first, let’s read all the documents into R!
During the last 2 days, we have created several output files we need for the statistical analysis, including (i) the frequency table sequenceData/8-final/zotutable.txt, (ii) the taxonomic ID file sequenceData/8-final/blastLineage.txt (let’s work on the BLAST taxonomy file for this tutorial), and (iii) the ZOTU sequence file sequenceData/8-final/zotus.fasta. Additionally, we have the sample metadata file sequenceData/0-metadata/metadata-COI-selected-updated.txt.
Note
copy-paste the code below in the R script underneath the code provided above.
##################
# READ DATA IN R #
##################
metaData <- read.table('../0-metadata/metadata-COI-selected-updated.txt', header = TRUE, sep = '\t', row.names = 1, check.names = FALSE, comment.char = '')
freqTable <- read.table('zotutable.txt', header = TRUE, sep = '\t', row.names = 1, check.names = FALSE, comment.char = '')
taxonomyTable <- read.table('blastLineage.txt', header = TRUE, sep = '\t', row.names = 1, check.names = FALSE, comment.char = '')
sequenceTable <- readDNAStringSet('zotus.fasta')
Warning
It is important to note that the files we just read into R should be seen as “linked together”, meaning that if we were to change one file due to some processing steps or analysis, we need to keep in mind to alter the other documents as well. The reason for this is that sample and sequence names need to be matching between the documents, otherwise errors will occur. So for example, if we remove a sequence from the frequency table, we need to remove the sequence in the ZOTU sequence file and taxonomic ID from the taxonomy table as well to keep the sequence lists between these three files identical.
3. Negative controls#
3.1 Investigating contamination#
One of the first pre-processing steps after generating the frequency table is to investigate what is observed in our negative control samples.
For the tutorial data set, we have three control samples that are labeled NTC_1, NTC_2, and NTC_3. To determine which sequences were present in our negative control samples and the read abundance of these signals, we can create a new column that is the sum of the three negative controls.
Note
For the tutorial data set, no metadata is provided to determine which control belongs to what sample or when and where they were collected. Therefore, summing them to a single sample would be fine in this instance. For your own project, adding negative control samples at various stages in your sample processing and barcoding the controls differently, will allow you to investigate contamination issues more in detail than covered in this tutorial.
#####################
# NEGATIVE CONTROLS #
#####################
# 1. find columns that contain NTC in header, 2. sum rows that contain NEG in header to column NEGSUM, 3. drop individual NEG columns
negColumns <- grep('NTC', names(freqTable), value = TRUE)
freqTable$NEGSUM <- rowSums(freqTable[negColumns])
freqTable <- freqTable[, !names(freqTable) %in% negColumns]
# 1. create list of index names (ZOTU numbers) for which freqTable$NEGSUM > 0, 2. print taxonomy of ZOTUs in list
negZOTUs <- rownames(freqTable)[which(freqTable$NEGSUM > 0)]
length(negZOTUs)
print(taxonomyTable[negZOTUs, ])
# 1. iterate over index names in negZOTUs list and print out the necessary values
for (x in negZOTUs) {
if (x %in% rownames(freqTable)) {
negValue <- freqTable[x, 'NEGSUM']
negValuePerc <- negValue / rowSums(freqTable[x, ]) * 100
rowMeanValue <- rowMeans(freqTable[x, ])
positiveDetections <- sum(freqTable[x, ] > 0)
positiveDetectionsPerc <- sum(freqTable[x, ] > 0) / nrow(freqTable) * 100
cat(x, 'value in NEG:', negValue, '% of ZOTU reads:', negValuePerc, 'mean # reads in sample:', rowMeanValue, '# +ve detections:', positiveDetections, '% +ve detections:', positiveDetectionsPerc, '\n')
}
}
Output
length(negZOTUs) [1] 99
print(taxonomyTable[negZOTUs, ]) kingdom phylum class order family genus species pident qcov zotu.1 Eukaryota Arthropoda Hexanauplia Cyclopoida Cyclopidae Cyclopidae_nan Cyclopidae_sp. 75.421 95 zotu.103 Eukaryota Arthropoda Malacostraca Decapoda Hippolytidae Hippolytidae_nan Hippolytidae_sp. 96.452 99 zotu.104 Eukaryota Arthropoda Arachnida Scorpiones Buthidae Centruroides Centruroides_vittatus 78.059 75 zotu.107 Eukaryota Arthropoda Malacostraca Amphipoda Amphipoda_nan Amphipoda_nan_nan Amphipoda_sp. 96.486 100 zotu.108 Eukaryota Annelida Polychaeta Eunicida Dorvilleidae Ophryotrocha Ophryotrocha_labronica 82.951 98 zotu.11 Eukaryota Arthropoda Malacostraca Decapoda Pilumnidae Pilumnidae_nan HK03 79.769 55 zotu.113 Eukaryota Bryozoa Gymnolaemata Ctenostomatida Vesiculariidae Amathia Amathia_sp. 85.256 99 zotu.116 Eukaryota Annelida Polychaeta Phyllodocida Syllidae Imajimaea Imajimaea_draculai 81.570 92 zotu.118 Eukaryota Annelida Polychaeta Terebellida Cirratulidae Dodecaceria Dodecaceria_sp. 90.735 100 zotu.1185 Eukaryota Chordata Ascidiacea Phlebobranchia Ascidiidae Ascidia pallida 85.809 97 zotu.119 Eukaryota Arthropoda Insecta Hymenoptera Ichneumonidae Baltazaria Baltazaria_sp. 74.809 83 zotu.1197 Eukaryota Arthropoda Thecostraca Balanomorpha Tetraclitidae Tetraclitella Tetraclitella_pilsbryi 100.000 100 zotu.12 Eukaryota Arthropoda Arachnida Araneae Hypochilidae Hypochilus Hypochilus_pococki 81.588 88 zotu.1207 Eukaryota Porifera Demospongiae Suberitida Suberitidae Suberitidae_nan Suberitidae_sp. 96.486 100 zotu.123 Eukaryota Arthropoda Malacostraca Amphipoda Podoceridae Podocerus brasiliensis 95.208 100 zotu.1274
NA NA zotu.13 Eukaryota Arthropoda Insecta Diptera Muscidae Muscina Muscina_pascuorum 80.412 93 zotu.135 Eukaryota Arthropoda Malacostraca Amphipoda Corophiidae Monocorophium acherusicum 97.125 100 zotu.139 Eukaryota Annelida Polychaeta Phyllodocida Syllidae Syllis Syllis_gracilis 80.822 93 zotu.143 Eukaryota Annelida Sipuncula Golfingiida Golfingiida_nan Golfingiida_nan_nan Golfingiida_sp. 100.000 100 zotu.158 Eukaryota Arthropoda Insecta Trichoptera Glossosomatidae Glossosomatidae_nan Glossosomatidae_gen. 82.156 86 zotu.161 Eukaryota Arthropoda Malacostraca Decapoda Porcellanidae Pisidia gordoni 99.038 99 zotu.171 Eukaryota Arthropoda Insecta Diptera Dolichopodidae Hydrophorus Hydrophorus_chrysologus 80.142 89 zotu.173 Eukaryota Arthropoda Insecta Lepidoptera Saturniidae Gynanisa Gynanisa_maja 84.076 99 zotu.175 Eukaryota Arthropoda Insecta Lepidoptera Nymphalidae Amphidecta Amphidecta_calliomma 79.221 98 zotu.177 Eukaryota Annelida Polychaeta Phyllodocida Hesionidae Micropodarke Micropodarke_sp. 100.000 100 zotu.18 Eukaryota Arthropoda Insecta Diptera Simuliidae Simuliidae_nan Simuliidae_sp. 77.076 96 zotu.183 Eukaryota Arthropoda Insecta Lepidoptera Riodinidae Mesosemia Mesosemia_gneris 83.387 99 zotu.188 Eukaryota Annelida Polychaeta Phyllodocida Hesionidae Platynereis HK04 100.000 100 zotu.193 Eukaryota Mollusca Bivalvia Galeommatida Galeommatidae Galeommatidae_nan HK01 100.000 100 zotu.2 Eukaryota Arthropoda Arachnida Opiliones Nemastomatidae Mitostoma Mitostoma_chrysomelas 76.596 89 zotu.20 Eukaryota Arthropoda Malacostraca Amphipoda Podoceridae Podocerus brasiliensis 93.610 100 zotu.207 Eukaryota Rhodophyta Florideophyceae Peyssonneliales Peyssonneliaceae Peyssonnelia Peyssonnelia_sp. 92.652 100 zotu.21 Eukaryota Arthropoda Diplopoda Polydesmida Xystodesmidae Brachoria Brachoria_laminata 76.654 81 zotu.211 Eukaryota Echinodermata Ophiuroidea Amphilepidida Ophiactidae Ophiactis Ophiactis_lymani 100.000 100 zotu.215 Eukaryota Arthropoda Remipedia Nectiopoda Speleonectidae Xibalbanus Xibalbanus_cozumelensis 75.779 92 zotu.22 Eukaryota Annelida Polychaeta Terebellida Cirratulidae Dodecaceria Dodecaceria_sp. 94.569 100 zotu.23 Eukaryota Arthropoda Malacostraca Amphipoda Podoceridae Podocerus brasiliensis 95.527 100 zotu.232 Eukaryota Porifera Demospongiae Suberitida Suberitidae Protosuberites Protosuberites_ectyoninus 98.403 100 zotu.244 Eukaryota Arthropoda Hexanauplia Harpacticoida Harpacticoida_nan Harpacticoida_nan_nan Harpacticoida_sp. 77.586 93 zotu.25 Eukaryota Arthropoda Malacostraca Decapoda Xanthidae Leptodius HK01 100.000 100 zotu.251 NA NA zotu.26 Eukaryota Arthropoda Malacostraca Amphipoda Amphipoda_nan Amphipoda_nan_nan Amphipoda_sp. 100.000 100 zotu.27 Eukaryota Bryozoa Gymnolaemata Cheilostomatida Schizoporellidae Schizoporella pungens 100.000 100 zotu.280 Eukaryota Bryozoa Gymnolaemata Cheilostomatida Microporellidae Microporella Microporella_sp. 82.274 96 zotu.29 Eukaryota Arthropoda Insecta Diptera Drosophilidae Zygothrica Zygothrica_sp. 79.655 92 zotu.3 Eukaryota Arthropoda Malacostraca Decapoda Alpheidae Athanas parvus 99.681 100 zotu.33 Eukaryota Arthropoda Insecta Hymenoptera Braconidae Aphidius Aphidius_smithi 77.193 90 zotu.330 Eukaryota Annelida Polychaeta Phyllodocida Syllidae Myrianida Myrianida_sp. 83.108 95 zotu.347 Eukaryota Arthropoda Insecta Coleoptera Cerambycidae Brothylus Brothylus_gemmulatus 77.287 99 zotu.36 Eukaryota Arthropoda Malacostraca Amphipoda Podoceridae Podocerus brasiliensis 95.847 100 zotu.389 Eukaryota Echinodermata Ophiuroidea Ophiuroidea_nan Ophiuroidea_nan_nan Ophiuroidea_nan_nan_nan HK02 90.032 99 zotu.4 Eukaryota Arthropoda Malacostraca Decapoda Hippolytidae Thor HK02 100.000 100 zotu.40 Eukaryota Echinodermata Ophiuroidea Amphilepidida Ophiactidae Ophiactis Ophiactis_lymani 98.718 99 zotu.42 Eukaryota Arthropoda Insecta Coleoptera Meloidae Meloe Meloe_proscarabaeus 73.841 96 zotu.441 Eukaryota Porifera Demospongiae Suberitida Suberitidae Suberitidae_nan Suberitidae_sp. 98.403 100 zotu.45 Eukaryota Arthropoda Hexanauplia Poecilostomatoida Clausidiidae Clausidium Clausidium_persiaensis 78.595 96 zotu.456 Eukaryota Annelida Polychaeta Eunicida Eunicidae Lysidice HK01 100.000 100 zotu.461 Eukaryota Arthropoda Insecta Diptera Ceratopogonidae Ceratopogonidae_nan Ceratopogonidae_sp. 79.322 94 zotu.47 Eukaryota Arthropoda Malacostraca Amphipoda Podoceridae Podocerus brasiliensis 93.291 100 zotu.474 Eukaryota Arthropoda Insecta Diptera Chironomidae Orthocladius Orthocladius_wetterensis 82.432 94 zotu.476 Eukaryota Arthropoda Insecta Lepidoptera Lycaenidae Cerautola Cerautola_miranda 82.143 98 zotu.5 Eukaryota Annelida Clitellata Crassiclitellata Megascolecidae Metaphire Metaphire_tosaensis 82.119 96 zotu.504 Eukaryota Annelida Polychaeta Terebellida Terebellidae Lysilla Lysilla_pacifica 89.776 100 zotu.522 Eukaryota Annelida Polychaeta Terebellida Cirratulidae Cirratulidae_nan Cirratulidae_sp. 80.479 94 zotu.533 Eukaryota Arthropoda Insecta Lepidoptera Lepidoptera_nan Lepidoptera_nan_nan Lepidoptera_sp. 83.972 90 zotu.541 Eukaryota Platyhelminthes Rhabditophora Polycladida Polycladida_nan Polycladida_nan_nan HK07 92.557 99 zotu.55 Eukaryota Annelida Polychaeta Eunicida Eunicidae Eunicidae_nan HK01 100.000 100 zotu.56 Eukaryota Arthropoda Hexanauplia Hexanauplia_nan Hexanauplia_nan_nan Hexanauplia_nan_nan_nan Hexanauplia_sp. 86.538 99 zotu.567 Eukaryota Arthropoda Insecta Lepidoptera Mimallonidae Pamea Pamea_perostia 81.208 95 zotu.58 Eukaryota Cnidaria Hydrozoa Leptothecata Clytiidae Clytia Clytia_sp. 95.498 99 zotu.59 Eukaryota Arthropoda Malacostraca Decapoda Pilumnidae Pilumnidae_nan HK03 79.191 55 zotu.6 Eukaryota Arthropoda Insecta Diptera Chironomidae Chironomidae_nan Orthocladiinae_sp. 78.777 88 zotu.605 Eukaryota Cnidaria Hydrozoa Leptothecata Clytiidae Clytia Clytia_sp. 97.764 100 zotu.607 Eukaryota Annelida Polychaeta Terebellida Cirratulidae Dodecaceria Dodecaceria_sp. 98.083 100 zotu.61 Eukaryota Arthropoda Thecostraca Balanomorpha Balanidae Amphibalanus Amphibalanus_reticulatus 100.000 100 zotu.616 Eukaryota Cnidaria Hydrozoa Anthoathecata Pandeidae Amphinema Amphinema_sp. 97.115 99 zotu.62 Eukaryota Annelida Polychaeta Spionida Spionidae Spionidae_nan HK01 100.000 100 zotu.658 Eukaryota Arthropoda Insecta Zygentoma Lepidotrichidae Tricholepidion Tricholepidion_gertschi 80.769 90 zotu.66 Eukaryota Chordata Mammalia Eulipotyphla Soricidae Blarina Blarina_brevicauda 80.128 50 zotu.696 Eukaryota Porifera Demospongiae Suberitida Suberitida_nan Terpios Terpios_gelatinosus 100.000 100 zotu.7 Eukaryota Annelida Polychaeta Phyllodocida Nereididae Platynereis Platynereis_dumerilii 84.158 96 zotu.72 Eukaryota Arthropoda Insecta Lepidoptera Tortricidae Eucosma Eucosma_sp. 80.592 97 zotu.75 Eukaryota Cnidaria Hydrozoa Leptothecata Eirenidae Eutima Eutima_mira 86.262 100 zotu.751 Eukaryota Rhodophyta Florideophyceae Corallinales Lithophyllaceae Amphiroa Amphiroa_beauvoisii 89.145 97 zotu.76 Eukaryota Annelida Polychaeta Eunicida Dorvilleidae Dorvillea HK01 100.000 100 zotu.785 Eukaryota Porifera Demospongiae Suberitida Suberitidae Protosuberites Protosuberites_mereui 100.000 100 zotu.79 Eukaryota Annelida Polychaeta Phyllodocida Hesionidae Syllidia Syllidia_sp. 100.000 100 zotu.799 Eukaryota Porifera Demospongiae Suberitida Suberitidae Protosuberites Protosuberites_denhartogi 97.444 100 zotu.8 Eukaryota Annelida Polychaeta Phyllodocida Nereididae Platynereis HK01 100.000 100 zotu.81 Eukaryota Arthropoda Hexanauplia Harpacticoida Tisbidae Tisbe Tisbe_sp. 78.758 98 zotu.836 Eukaryota Annelida Polychaeta Polychaeta_nan Arenicolidae Arenicola Arenicola 83.601 99 zotu.85 Eukaryota Arthropoda Hexanauplia Harpacticoida Ameiridae Pseudameira Pseudameira_crassicornis 74.839 99 zotu.867 Eukaryota Annelida Polychaeta Spionida Spionidae Pygospio Pygospio_elegans 79.211 89 zotu.886 Eukaryota Cnidaria Anthozoa Scleractinia Dendrophylliidae Duncanopsammia Duncanopsammia_peltata 76.000 95 zotu.9 Eukaryota Annelida Polychaeta Phyllodocida Nereididae Platynereis HK01 100.000 100 zotu.90 Eukaryota Arthropoda Malacostraca Decapoda Porcellanidae Pisidia gordoni 100.000 100 zotu.95 Eukaryota Arthropoda Arachnida Trombidiformes Rhagidiidae Rhagidiidae_nan Rhagidiidae_sp. 79.612 98 zotu.98 Eukaryota Arthropoda Arachnida Sarcoptiformes Oppiidae Oppiidae_nan Oppiidae_sp. 79.741 73
zotu.1 value in NEG: 23 % of ZOTU reads: 0.01085197 mean # reads in sample: 8477.72 # +ve detections: 25 % +ve detections: 1.858736 zotu.103 value in NEG: 1 % of ZOTU reads: 0.03518649 mean # reads in sample: 113.68 # +ve detections: 8 % +ve detections: 0.5947955 zotu.104 value in NEG: 1 % of ZOTU reads: 0.03447087 mean # reads in sample: 116.04 # +ve detections: 7 % +ve detections: 0.5204461 zotu.107 value in NEG: 1 % of ZOTU reads: 0.03932363 mean # reads in sample: 101.72 # +ve detections: 8 % +ve detections: 0.5947955 zotu.108 value in NEG: 1 % of ZOTU reads: 0.03773585 mean # reads in sample: 106 # +ve detections: 16 % +ve detections: 1.189591 zotu.11 value in NEG: 3 % of ZOTU reads: 0.007677936 mean # reads in sample: 1562.92 # +ve detections: 15 % +ve detections: 1.115242 zotu.113 value in NEG: 1 % of ZOTU reads: 0.04395604 mean # reads in sample: 91 # +ve detections: 8 % +ve detections: 0.5947955 zotu.116 value in NEG: 1 % of ZOTU reads: 0.04401408 mean # reads in sample: 90.88 # +ve detections: 9 % +ve detections: 0.669145 zotu.118 value in NEG: 3 % of ZOTU reads: 0.1345895 mean # reads in sample: 89.16 # +ve detections: 14 % +ve detections: 1.040892 zotu.1185 value in NEG: 1 % of ZOTU reads: 2.5 mean # reads in sample: 1.6 # +ve detections: 11 % +ve detections: 0.8178439 zotu.119 value in NEG: 1 % of ZOTU reads: 0.04531038 mean # reads in sample: 88.28 # +ve detections: 4 % +ve detections: 0.2973978 zotu.1197 value in NEG: 3 % of ZOTU reads: 3.896104 mean # reads in sample: 3.08 # +ve detections: 16 % +ve detections: 1.189591 zotu.12 value in NEG: 7 % of ZOTU reads: 0.01907357 mean # reads in sample: 1468 # +ve detections: 19 % +ve detections: 1.412639 zotu.1207 value in NEG: 1 % of ZOTU reads: 1.149425 mean # reads in sample: 3.48 # +ve detections: 10 % +ve detections: 0.7434944 zotu.123 value in NEG: 1 % of ZOTU reads: 0.0463392 mean # reads in sample: 86.32 # +ve detections: 8 % +ve detections: 0.5947955 zotu.1274 value in NEG: 1 % of ZOTU reads: 1.219512 mean # reads in sample: 3.28 # +ve detections: 2 % +ve detections: 0.1486989 zotu.13 value in NEG: 4 % of ZOTU reads: 0.01303229 mean # reads in sample: 1227.72 # +ve detections: 12 % +ve detections: 0.8921933 zotu.135 value in NEG: 1 % of ZOTU reads: 0.05083884 mean # reads in sample: 78.68 # +ve detections: 9 % +ve detections: 0.669145 zotu.139 value in NEG: 5 % of ZOTU reads: 0.2503756 mean # reads in sample: 79.88 # +ve detections: 17 % +ve detections: 1.263941 zotu.143 value in NEG: 2 % of ZOTU reads: 0.1144165 mean # reads in sample: 69.92 # +ve detections: 12 % +ve detections: 0.8921933 zotu.158 value in NEG: 2 % of ZOTU reads: 0.1273074 mean # reads in sample: 62.84 # +ve detections: 14 % +ve detections: 1.040892 zotu.161 value in NEG: 1 % of ZOTU reads: 0.07173601 mean # reads in sample: 55.76 # +ve detections: 11 % +ve detections: 0.8178439 zotu.171 value in NEG: 1 % of ZOTU reads: 0.08038585 mean # reads in sample: 49.76 # +ve detections: 9 % +ve detections: 0.669145 zotu.173 value in NEG: 4 % of ZOTU reads: 0.3325021 mean # reads in sample: 48.12 # +ve detections: 20 % +ve detections: 1.486989 zotu.175 value in NEG: 1 % of ZOTU reads: 0.08368201 mean # reads in sample: 47.8 # +ve detections: 11 % +ve detections: 0.8178439 zotu.177 value in NEG: 2 % of ZOTU reads: 0.1631321 mean # reads in sample: 49.04 # +ve detections: 8 % +ve detections: 0.5947955 zotu.18 value in NEG: 2 % of ZOTU reads: 0.01051359 mean # reads in sample: 760.92 # +ve detections: 20 % +ve detections: 1.486989 zotu.183 value in NEG: 2 % of ZOTU reads: 0.1858736 mean # reads in sample: 43.04 # +ve detections: 12 % +ve detections: 0.8921933 zotu.188 value in NEG: 1 % of ZOTU reads: 0.09551098 mean # reads in sample: 41.88 # +ve detections: 7 % +ve detections: 0.5204461 zotu.193 value in NEG: 1 % of ZOTU reads: 0.09569378 mean # reads in sample: 41.8 # +ve detections: 5 % +ve detections: 0.3717472 zotu.2 value in NEG: 3 % of ZOTU reads: 0.001785364 mean # reads in sample: 6721.32 # +ve detections: 22 % +ve detections: 1.635688 zotu.20 value in NEG: 6 % of ZOTU reads: 0.03762699 mean # reads in sample: 637.84 # +ve detections: 12 % +ve detections: 0.8921933 zotu.207 value in NEG: 1 % of ZOTU reads: 0.1122334 mean # reads in sample: 35.64 # +ve detections: 13 % +ve detections: 0.9665428 zotu.21 value in NEG: 1 % of ZOTU reads: 0.006319914 mean # reads in sample: 632.92 # +ve detections: 21 % +ve detections: 1.561338 zotu.211 value in NEG: 2 % of ZOTU reads: 0.2118644 mean # reads in sample: 37.76 # +ve detections: 8 % +ve detections: 0.5947955 zotu.215 value in NEG: 2 % of ZOTU reads: 0.2229654 mean # reads in sample: 35.88 # +ve detections: 8 % +ve detections: 0.5947955 zotu.22 value in NEG: 10 % of ZOTU reads: 0.06314726 mean # reads in sample: 633.44 # +ve detections: 24 % +ve detections: 1.784387 zotu.23 value in NEG: 2 % of ZOTU reads: 0.01288494 mean # reads in sample: 620.88 # +ve detections: 10 % +ve detections: 0.7434944 zotu.232 value in NEG: 1 % of ZOTU reads: 0.1336898 mean # reads in sample: 29.92 # +ve detections: 12 % +ve detections: 0.8921933 zotu.244 value in NEG: 1 % of ZOTU reads: 0.1519757 mean # reads in sample: 26.32 # +ve detections: 6 % +ve detections: 0.4460967 zotu.25 value in NEG: 1 % of ZOTU reads: 0.007007217 mean # reads in sample: 570.84 # +ve detections: 21 % +ve detections: 1.561338 zotu.251 value in NEG: 1 % of ZOTU reads: 0.1519757 mean # reads in sample: 26.32 # +ve detections: 3 % +ve detections: 0.2230483 zotu.26 value in NEG: 1 % of ZOTU reads: 0.007456566 mean # reads in sample: 536.44 # +ve detections: 14 % +ve detections: 1.040892 zotu.27 value in NEG: 7 % of ZOTU reads: 0.0519442 mean # reads in sample: 539.04 # +ve detections: 24 % +ve detections: 1.784387 zotu.280 value in NEG: 4 % of ZOTU reads: 0.754717 mean # reads in sample: 21.2 # +ve detections: 16 % +ve detections: 1.189591 zotu.29 value in NEG: 2 % of ZOTU reads: 0.01520681 mean # reads in sample: 526.08 # +ve detections: 14 % +ve detections: 1.040892 zotu.3 value in NEG: 5 % of ZOTU reads: 0.003639222 mean # reads in sample: 5495.68 # +ve detections: 25 % +ve detections: 1.858736 zotu.33 value in NEG: 1 % of ZOTU reads: 0.008809796 mean # reads in sample: 454.04 # +ve detections: 10 % +ve detections: 0.7434944 zotu.330 value in NEG: 2 % of ZOTU reads: 0.4424779 mean # reads in sample: 18.08 # +ve detections: 5 % +ve detections: 0.3717472 zotu.347 value in NEG: 9 % of ZOTU reads: 2.472527 mean # reads in sample: 14.56 # +ve detections: 8 % +ve detections: 0.5947955 zotu.36 value in NEG: 1 % of ZOTU reads: 0.008963786 mean # reads in sample: 446.24 # +ve detections: 8 % +ve detections: 0.5947955 zotu.389 value in NEG: 2 % of ZOTU reads: 0.6369427 mean # reads in sample: 12.56 # +ve detections: 6 % +ve detections: 0.4460967 zotu.4 value in NEG: 6 % of ZOTU reads: 0.004914126 mean # reads in sample: 4883.88 # +ve detections: 18 % +ve detections: 1.33829 zotu.40 value in NEG: 7 % of ZOTU reads: 0.07356805 mean # reads in sample: 380.6 # +ve detections: 16 % +ve detections: 1.189591 zotu.42 value in NEG: 1 % of ZOTU reads: 0.009761812 mean # reads in sample: 409.76 # +ve detections: 16 % +ve detections: 1.189591 zotu.441 value in NEG: 16 % of ZOTU reads: 5.714286 mean # reads in sample: 11.2 # +ve detections: 24 % +ve detections: 1.784387 zotu.45 value in NEG: 4 % of ZOTU reads: 0.0518605 mean # reads in sample: 308.52 # +ve detections: 16 % +ve detections: 1.189591 zotu.456 value in NEG: 1 % of ZOTU reads: 0.4201681 mean # reads in sample: 9.52 # +ve detections: 11 % +ve detections: 0.8178439 zotu.461 value in NEG: 2 % of ZOTU reads: 0.6825939 mean # reads in sample: 11.72 # +ve detections: 11 % +ve detections: 0.8178439 zotu.47 value in NEG: 1 % of ZOTU reads: 0.01359804 mean # reads in sample: 294.16 # +ve detections: 7 % +ve detections: 0.5204461 zotu.474 value in NEG: 1 % of ZOTU reads: 0.4739336 mean # reads in sample: 8.44 # +ve detections: 7 % +ve detections: 0.5204461 zotu.476 value in NEG: 3 % of ZOTU reads: 1.388889 mean # reads in sample: 8.64 # +ve detections: 16 % +ve detections: 1.189591 zotu.5 value in NEG: 7 % of ZOTU reads: 0.009387531 mean # reads in sample: 2982.68 # +ve detections: 23 % +ve detections: 1.710037 zotu.504 value in NEG: 1 % of ZOTU reads: 0.456621 mean # reads in sample: 8.76 # +ve detections: 7 % +ve detections: 0.5204461 zotu.522 value in NEG: 1 % of ZOTU reads: 0.4950495 mean # reads in sample: 8.08 # +ve detections: 10 % +ve detections: 0.7434944 zotu.533 value in NEG: 1 % of ZOTU reads: 0.5813953 mean # reads in sample: 6.88 # +ve detections: 9 % +ve detections: 0.669145 zotu.541 value in NEG: 2 % of ZOTU reads: 1.176471 mean # reads in sample: 6.8 # +ve detections: 12 % +ve detections: 0.8921933 zotu.55 value in NEG: 1 % of ZOTU reads: 0.01510346 mean # reads in sample: 264.84 # +ve detections: 9 % +ve detections: 0.669145 zotu.56 value in NEG: 1 % of ZOTU reads: 0.01590331 mean # reads in sample: 251.52 # +ve detections: 17 % +ve detections: 1.263941 zotu.567 value in NEG: 1 % of ZOTU reads: 0.6410256 mean # reads in sample: 6.24 # +ve detections: 8 % +ve detections: 0.5947955 zotu.58 value in NEG: 3 % of ZOTU reads: 0.04970179 mean # reads in sample: 241.44 # +ve detections: 14 % +ve detections: 1.040892 zotu.59 value in NEG: 1 % of ZOTU reads: 0.01670844 mean # reads in sample: 239.4 # +ve detections: 7 % +ve detections: 0.5204461 zotu.6 value in NEG: 5 % of ZOTU reads: 0.008204388 mean # reads in sample: 2437.72 # +ve detections: 22 % +ve detections: 1.635688 zotu.605 value in NEG: 1 % of ZOTU reads: 0.6849315 mean # reads in sample: 5.84 # +ve detections: 11 % +ve detections: 0.8178439 zotu.607 value in NEG: 1 % of ZOTU reads: 0.8333333 mean # reads in sample: 4.8 # +ve detections: 5 % +ve detections: 0.3717472 zotu.61 value in NEG: 2 % of ZOTU reads: 0.03589375 mean # reads in sample: 222.88 # +ve detections: 22 % +ve detections: 1.635688 zotu.616 value in NEG: 1 % of ZOTU reads: 0.729927 mean # reads in sample: 5.48 # +ve detections: 11 % +ve detections: 0.8178439 zotu.62 value in NEG: 1 % of ZOTU reads: 0.0183925 mean # reads in sample: 217.48 # +ve detections: 15 % +ve detections: 1.115242 zotu.658 value in NEG: 2 % of ZOTU reads: 1.754386 mean # reads in sample: 4.56 # +ve detections: 14 % +ve detections: 1.040892 zotu.66 value in NEG: 1 % of ZOTU reads: 0.01907305 mean # reads in sample: 209.72 # +ve detections: 6 % +ve detections: 0.4460967 zotu.696 value in NEG: 3 % of ZOTU reads: 2.238806 mean # reads in sample: 5.36 # +ve detections: 12 % +ve detections: 0.8921933 zotu.7 value in NEG: 4 % of ZOTU reads: 0.006447245 mean # reads in sample: 2481.68 # +ve detections: 20 % +ve detections: 1.486989 zotu.72 value in NEG: 1 % of ZOTU reads: 0.02071251 mean # reads in sample: 193.12 # +ve detections: 6 % +ve detections: 0.4460967 zotu.75 value in NEG: 2 % of ZOTU reads: 0.04334634 mean # reads in sample: 184.56 # +ve detections: 12 % +ve detections: 0.8921933 zotu.751 value in NEG: 1 % of ZOTU reads: 1.176471 mean # reads in sample: 3.4 # +ve detections: 13 % +ve detections: 0.9665428 zotu.76 value in NEG: 2 % of ZOTU reads: 0.04344049 mean # reads in sample: 184.16 # +ve detections: 12 % +ve detections: 0.8921933 zotu.785 value in NEG: 8 % of ZOTU reads: 6.349206 mean # reads in sample: 5.04 # +ve detections: 19 % +ve detections: 1.412639 zotu.79 value in NEG: 1 % of ZOTU reads: 0.01793722 mean # reads in sample: 223 # +ve detections: 24 % +ve detections: 1.784387 zotu.799 value in NEG: 2 % of ZOTU reads: 2.5 mean # reads in sample: 3.2 # +ve detections: 3 % +ve detections: 0.2230483 zotu.8 value in NEG: 10 % of ZOTU reads: 0.01740765 mean # reads in sample: 2297.84 # +ve detections: 25 % +ve detections: 1.858736 zotu.81 value in NEG: 1 % of ZOTU reads: 0.02248707 mean # reads in sample: 177.88 # +ve detections: 16 % +ve detections: 1.189591 zotu.836 value in NEG: 1 % of ZOTU reads: 1.234568 mean # reads in sample: 3.24 # +ve detections: 8 % +ve detections: 0.5947955 zotu.85 value in NEG: 1 % of ZOTU reads: 0.02480159 mean # reads in sample: 161.28 # +ve detections: 19 % +ve detections: 1.412639 zotu.867 value in NEG: 1 % of ZOTU reads: 0.8547009 mean # reads in sample: 4.68 # +ve detections: 8 % +ve detections: 0.5947955 zotu.886 value in NEG: 1 % of ZOTU reads: 1.388889 mean # reads in sample: 2.88 # +ve detections: 10 % +ve detections: 0.7434944 zotu.9 value in NEG: 20 % of ZOTU reads: 0.0434537 mean # reads in sample: 1841.04 # +ve detections: 25 % +ve detections: 1.858736 zotu.90 value in NEG: 1 % of ZOTU reads: 0.0272257 mean # reads in sample: 146.92 # +ve detections: 14 % +ve detections: 1.040892 zotu.95 value in NEG: 5 % of ZOTU reads: 0.1363884 mean # reads in sample: 146.64 # +ve detections: 23 % +ve detections: 1.710037 zotu.98 value in NEG: 3 % of ZOTU reads: 0.08846948 mean # reads in sample: 135.64 # +ve detections: 11 % +ve detections: 0.8178439
The output shows that we have 99 sequences that had a positive detection in the negative control samples. These positive detections in the negative control samples included some of the most abundant sequences in the data set, including the top 9 ZOTU sequences. Looking at the read abundance, however, shows that the signal is quite weak compared to the actual samples.
Important
While the read count of the negative detections is low compared to the actual samples, no information is provided in the metadata on how the negative control samples were added to the sequencing library. If the negative control samples were spiked into the library, rather than pooled equimolarly, the low read count will be an artefact of the lab work and not a true reflection of the level of contamination observed in the data set.
3.2 Dealing with contaminants#
There are a variety of ways on how you can process contamination in your sequence data. All of them can be justified, but the method should be clearly stated in the manuscript, as it might impact downstream results and interpretation of the analysis.
3.2.1 Strict filtering#
One of the most conserved ways to deal with contamination is to remove all sequences with a positive detection in the negative controls, plus remove any single detections with a value equal or lower than the highest read count observed in the negative controls. For this tutorial data set, such an approach would probably not be advised, as you will remove all the high-abundant sequences from the analysis. However, below is the code on how to conduct such a filtering approach.
# 1. determine the maximum number of reads observed in the negative controls, 2. set detection of threshold or lower to 0, 3. remove contaminant sequences from data frame
negThreshold <- max(freqTable$NEGSUM)
freqTable.strictFilter <- as.data.frame(apply(freqTable, 2, function(x) ifelse(x < negThreshold, 0, x)))
freqTable.strictFilter <- freqTable.strictFilter[!(rownames(freqTable.strictFilter) %in% negZOTUs), ]
sum(freqTable)
sum(freqTable.strictFilter)
sum(freqTable.strictFilter) / sum(freqTable) * 100
colSums(freqTable.strictFilter)
min(colSums(freqTable.strictFilter)[colSums(freqTable.strictFilter) != 0], na.rm = TRUE)
This filtering approach not only removed 99 ZOTUs from the data set, but also reduced the total read count from 2,195,816 to 801,517 (36.5%). While the read count has been reduced significantly, no sample dropout was observed and the minimal total read count for a sample is 9,803.
3.2.2 Relaxed filtering#
A relaxed alternative to deal with contamination is to set a threshold value for keeping a “true” detection based on the read abundance in the negative controls. This value could either be the read abundance in the negative controls or a multiplier of this value. For this example, let’s set positive detections in the samples to 0 if they do not reach 10 times the number of reads observed in the negative controls.
# 1. initialise new df, 2. for loop to set all values less than 10 x the negative control value to 0
freqTable.relaxedFilter <- freqTable
for (negZOTU in negZOTUs) {
condition <- freqTable.relaxedFilter[negZOTU, 'NEGSUM'] * 10
freqTable.relaxedFilter[negZOTU, -1] <- ifelse(freqTable.relaxedFilter[negZOTU, -1] > condition, freqTable.relaxedFilter[negZOTU, -1], 0)
}
sum(freqTable)
sum(freqTable.relaxedFilter)
sum(freqTable.relaxedFilter) / sum(freqTable) * 100
rownames(freqTable.relaxedFilter)[which(rowSums(freqTable.relaxedFilter) == 0)]
The relaxed approach only reduced total read count from 2,195,816 to 2,191,995 (99.8%) and removed a single ZOTU (zotu.1185) from the data set.
3.2.3 decontam R package#
The final option we will explore in this tutorial to deal with contamination in metabarcoding data is the decontam R package introduced in the paper Davis et al., 2018. This package provides a statistical test to identify contaminants based on two widely reproduced patterns, including (i) contaminants appear at higher frequencies in low-concentration samples and (ii) are often found in negative controls.
Note
It should be noted that this approach is more suitable when DNA concentrations, measured by fluorescent intensity, are available for all samples in the metadata file. In this case, the frequency method can be used, rather than the more restrictive prevalence method we will be using today.
3.2.3.1 Phyloseq import#
The decontam R package is linked to the phyloseq R package. Hence, before running decontam, we need to import our files into phyloseq objects. Additionally, we need to read in the frequency table again, as we have altered this data frame in the code above. For decontam, it is important that we keep all the negative control samples separately.
# 1. read frequency table into memory again
freqTable <- read.table('zotutable.txt', header = TRUE, sep = '\t', row.names = 1, check.names = FALSE, comment.char = '')
# 1. import the frequency table into phyloseq, 2. import the metadata table into phyloseq, 3. merge phyloseq objects
OTU = otu_table(freqTable, taxa_are_rows = TRUE)
META = sample_data(metaData)
physeq = merge_phyloseq(OTU, META)
physeq
Output
> physeq
phyloseq-class experiment-level object
otu_table() OTU Table: [ 1345 taxa and 27 samples ]
sample_data() Sample Data: [ 27 samples by 32 sample variables ]
3.2.3.2 Plot total read count#
After importing the dataframes into phyloseq objects, we can have a look at the library sizes (i.e., the number of reads in each sample), as a function of whether that sample was a true sample or a negative control
# 1. transform sample_data to ggplot-friendly data frame, 2. sum reads per sample, 3. order on total read count, 4. set index column for plotting, 5. plot data in ggplot
decontam.df <- as.data.frame(sample_data(physeq))
decontam.df$LibrarySize <- sample_sums(physeq)
decontam.df <- decontam.df[order(decontam.df$LibrarySize),]
decontam.df$Index <- seq(nrow(decontam.df))
ggplot(data = decontam.df, aes(x = Index, y = LibrarySize, color = island)) + geom_point()
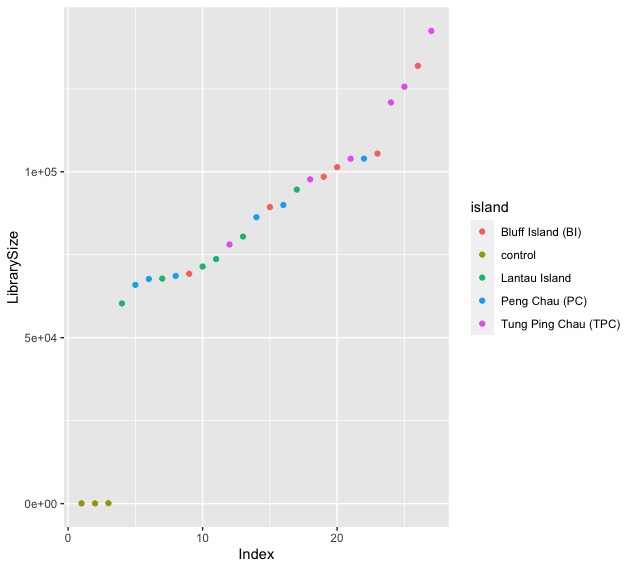
Fig. 32 : The library size (number of reads) for each sample. Note the difference between the negative controls and the true samples.#
The library sizes of the true samples fall between ~50,000 to ~140,000 reads without any outliers that fall within the range of the negative controls. The negative control samples, on the other hand, have far fewer reads compared to true samples, i.e., ~100 reads.
3.2.3.3 Determine contaminants#
The next step is to create an additional column in our metadata phyloseq object that holds the negative control sample information as a logical variable, with TRUE for negative control samples. Once the column is created, we can run the isContaminant function with method = 'prevalence' to identify the contaminants according to the decontam R package. We will set the threshold = 0.5, which will allow decontam to identify contaminants as all sequences that are more prevalent in negative control samples than in true samples. The isContaminant function will return a dataframe with several columns, the most importnat being $p containing the probability for classifying contaminants and $contaminant which contains TRUE/FALSE classification values with TRUE indicating that the statistical evidence that the associated sequence feature is a contaminant exceeds the user-settable threshold (in our case 0.5.)
# 1. create new column with logical variables, 2. determine contaminants, 3. return summary table, 4. display first entries
sample_data(physeq)$is.neg <- sample_data(physeq)$island == 'control'
contamdf.prev <- isContaminant(physeq, method = 'prevalence', neg = 'is.neg', threshold = 0.5)
table(contamdf.prev$contaminant)
head(which(contamdf.prev$contaminant))
Output
> table(contamdf.prev$contaminant)
FALSE TRUE
1297 48
> head(which(contamdf.prev$contaminant))
[1] 36 47 80 147 180 202
The output shows that decontam identified 48 sequences as contamination. Interestingly, the highly abundant sequences are not identified as contaminants, as the prevalence in true samples was too high compared to the negative controls.
3.2.3.4 Plot prevalence#
We can plot the sequences as a function of the prevalence in negative controls and true samples. When coloured by contaminant sequences, we can investigate the results of decontam.
# 1. transform phyloseq object to presence-absence, 2. subset negative control samples, 3. subset true samples, 4. make data.frame of prevalence in positive and negative samples, 5. plot in ggplot
physeq.pa <- transform_sample_counts(physeq, function(abund) 1*(abund>0))
physeq.pa.neg <- prune_samples(sample_data(physeq.pa)$island == 'control', physeq.pa)
physeq.pa.pos <- prune_samples(sample_data(physeq.pa)$island != 'control', physeq.pa)
df.pa <- data.frame(pa.pos = taxa_sums(physeq.pa.pos), pa.neg = taxa_sums(physeq.pa.neg), contaminant = contamdf.prev$contaminant)
ggplot(data = df.pa, aes(x = pa.neg, y = pa.pos, color = contaminant)) + geom_point() + xlab('Prevalence (Negative controls)') + ylab('Prevalence (True samples)')
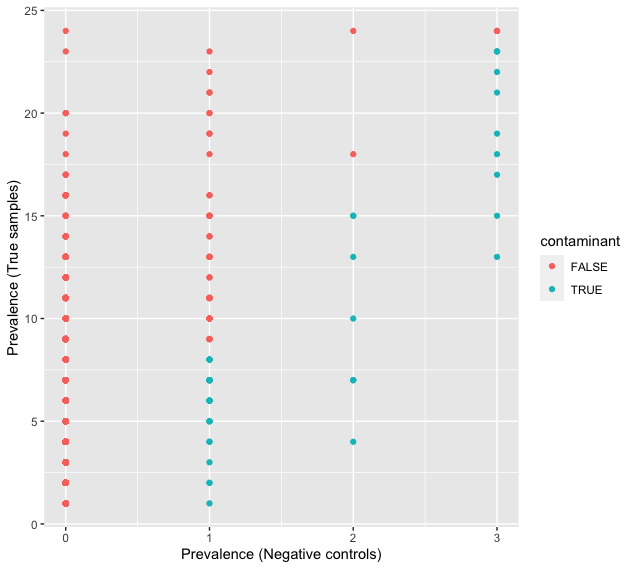
Fig. 33 : The prevalence of detections of sequences in negative controls and true samples#
3.2.3.5 Remove contaminants#
The final step is to subset the frequency table and sequence list, whereby we remove the contaminants as identified by the decontam package.
# 1. identify contaminants, 2. remove contaminants from frequence table, 3. remove contaminants from sequence list
contamSeq <- row.names(contamdf.prev)[with(contamdf.prev, contaminant %in% c(TRUE))]
freqTable.clean <- freqTable[!row.names(freqTable) %in% contamSeq, ]
sequenceTable.clean <- sequenceTable[rownames(freqTable.clean)]
sum(freqTable)
sum(freqTable.clean)
sum(freqTable.clean) / sum(freqTable) * 100
colSums(freqTable.clean)
The decontam package reduced the read count in our frequency table from 2,195,816 to 1,786,221 (81.3%) reads. Similarly as before, no sample drop out has occurred during this filtering process. Note that even the negative control samples still contain reads.
4. Abundance filtering#
One frequently included processing step for metabarcoding data is to remove low abundant detections, i.e., detections that are made up by a single or a few reads. This pre-processing step is conducted to minimise the risk of cross-contamination between samples or to eliminate the effect of tag jumps. Tag jumps are sequences that have been assigned to the wrong sample, i.e., contain the wrong tags or barcodes. You can read more about this in this paper by Schnell et al., 2015. The threshold set for abundance filtering is user-defined and can be to remove singleton detection or based on the total abundance of reads for a ZOTU sequence.
Since we provided the code on abundance filtering based on a threshold value during section 3.2.1 Strict filtering, we’ll now opt to filter out all singleton detections from the dataframe. For this tutorial, we’ll work on the dataframes created after running the decontam functions.
#############
# ABUNDANCE #
#############
# 1. set all values lower than 2 to 0, 2. check if lowest value in dataframe is 2, excluding 0
freqTable.clean[freqTable.clean < 2] <- 0
min(freqTable.clean[freqTable.clean != 0], na.rm = TRUE)
Output
> min(freqTable.clean[freqTable.clean != 0], na.rm = TRUE)
[1] 2
5. Low confidence tax IDs#
Another pre-processing step certain metabarcoding projects undertake is the removal of low-confidence taxonomic IDs. Removal of low-confidence taxonomic IDs and sequences without a taxonomic ID allows for curation of artefact sequences and taxon-dependent analyses. This approach is most commonly used in single- or multi-marker targeted metabarcoding projects, rather than a project utilizing one or more “universal” primer sets. The difference in approach is due to the completeness of the reference database, e.g., fish or mammal reference databases are much more complete compared to phytoplankton databases. For our dataset, we can remove the ZOTU sequences for which no BLAST hit was achieved.
#########################
# LOW CONFIDENCE TAX ID #
#########################
# 1. find ZOTUs which do not have a taxonomic ID at order level, 2. remove this list from the frequency table
lowConfidenceTax <- rownames(taxonomyTable)[which(is.na(taxonomyTable$order))]
freqTable.lowConfidenceTax <- freqTable[!(rownames(freqTable) %in% lowConfidenceTax), ]
length(lowConfidenceTax)
lowConfidenceTax
sum(freqTable)
sum(freqTable.lowConfidenceTax)
sum(freqTable.lowConfidenceTax) / sum(freqTable) * 100
colSums(freqTable.lowConfidenceTax)
Output
> length(lowConfidenceTax)
[1] 58
> lowConfidenceTax
[1] "zotu.110" "zotu.166" "zotu.209" "zotu.227" "zotu.251" "zotu.257" "zotu.303" "zotu.317" "zotu.321" "zotu.339" "zotu.362" "zotu.365" "zotu.437" "zotu.459" "zotu.532"
[16] "zotu.543" "zotu.589" "zotu.602" "zotu.622" "zotu.624" "zotu.627" "zotu.657" "zotu.693" "zotu.717" "zotu.726" "zotu.756" "zotu.810" "zotu.891" "zotu.909" "zotu.919"
[31] "zotu.932" "zotu.941" "zotu.979" "zotu.997" "zotu.1024" "zotu.1067" "zotu.1070" "zotu.1076" "zotu.1080" "zotu.1082" "zotu.1091" "zotu.1093" "zotu.1104" "zotu.1111" "zotu.1124"
[46] "zotu.1127" "zotu.1145" "zotu.1160" "zotu.1180" "zotu.1184" "zotu.1223" "zotu.1232" "zotu.1244" "zotu.1249" "zotu.1274" "zotu.1280" "zotu.1290" "zotu.1302"
> sum(freqTable)
[1] 2195816
> sum(freqTable.lowConfidenceTax)
[1] 2181081
> sum(freqTable.lowConfidenceTax) / sum(freqTable) * 100
[1] 99.32895
For the tutorial data, removal of sequences without a BLAST hit removed 58 ZOTU sequences and reduced the total read count from 2,195,816 to 2,181,081 (99.3%) reads.
6. Artefact sequences#
During the bioinformatic pipeline, we have attempted to remove low-quality and artificial (chimeric) sequences. However, artefact sequences can and most likely will have persisted in our data set up until this stage. For example, we set the expected error threshold at 1 per read, allowing for artificial variation in the dataset. Denoising should have merged most of such artefacts, but algorithms are not perfect. Artefacts can also be brought in existence due to PCR amplification errors. While chimeras are one form, multiple other variants of PCR errors exist. See Potapov et al., 2017 for more information.
To further identify and remove artefact sequences from our data set, we can use the LULU algorithm from Froslev et al., 2017. This algorithm is based on co-occurrence patterns whereby an artefact or child sequence is identified and merged to its parent when the child sequence is only present in lower abundance in samples of the parent (you cannot create an artefact without it’s initial sequence) and the similarity between parent and child reaches a certain identity threshold.
Besides the frequency table, we need a “match list” for LULU to work. This match list is a simple BLAST search for the ZOTU sequences against themselves to identify the similarity value between the sequences. As we’ve worked with BLAST before during the taxonomy assignment, we can move past this quite quickly. A more complete set of instructions can be found on LULU’s GitHub repo. To generate the match list, we need (1) to create a BLAST database from the ZOTU sequences and (2) run a blastn search of the ZOTU sequences against the database. Note that this should be done in the Terminal
cd sequenceData/8-final
makeblastdb -in zotus.fasta -parse_seqids -dbtype nucl
blastn -db zotus.fasta -outfmt '6 qseqid sseqid pident' -out match_list.txt -qcov_hsp_perc 80 -perc_identity 84 -query zotus.fasta
The code above will have created the sequenceData/8-final/match_list.txt file, which we can view using the head command.
head -n 10 match_list.txt
Output
zotu.1 zotu.1 100.000
zotu.1 zotu.42 99.361
zotu.1 zotu.731 98.403
zotu.1 zotu.744 98.083
zotu.2 zotu.2 100.000
zotu.2 zotu.728 98.083
zotu.3 zotu.3 100.000
zotu.3 zotu.34 99.361
zotu.3 zotu.164 99.042
zotu.3 zotu.43 99.042
Next, we can import the match list into R and run the LULU algorithm using the R code below.
########
# LULU #
########
# 1. read in the BLAST results, 2. run the lulu algorithm, 3. determine the number of artefact sequences, 4. export new frequency table from LULU
matchlist <- read.table('match_list.txt', header = FALSE, as.is = TRUE, stringsAsFactors = FALSE)
curatedResult <- lulu(freqTable, matchlist)
curatedResult$discarded_count
freqTable.lulu <- curatedResult$curated_table
sum(freqTable)
sum(freqTable.lulu)
The LULU algorithm identified 369 additional artefact sequences and merged them with the parent, leaving 976 ZOTU sequences in the cleaned up frequency table. As LULU merges child sequences with their parent, no reads were lost during this analysis.
7. Rarefaction#
One final pre-processing step deals with differences in sequencing depth between samples. With the realisation that a higher read count could lead to a larger number of ZOTU sequences being detected, there has been much debate in the scientific community on how to deal with this issue. Some studies argue that it is essential to “level the playing field” and use the same number of sequences across all samples. This random subsampling of the data is called rarefaction. See Cameron et al., 2021 as an example. Other studies, on the other hand, argue vehemently against such an approach. For example, you can read McMurdie et al., 2014 for more on this topic. While this debate is still ongoing as far as I’m aware, I suggest you chose what suits you best and clearly state what and how you have processed your data.
There are three steps you can take to determine if rarefying your data might be necessary. The first is to determine if there is a significant difference in read depth between samples. The second is to determine if there is a correlation between sequencing depth and the number of ZOTU sequences in a sample. The third is to draw so-called rarefaction curves, which are similar to species accumulation curves, and determine if sufficient sequencing coverage is achieved for samples when the curves plateau, indicating all species or ZOTU sequences were detected in the samples.
7.1 Read distribution#
Let’s start with the first option. Here, we can plot the read distribution for samples and ZOTU sequences as a visualisation. We can plot the y-axis both without transformation and log-transformed.
###############
# RAREFACTION #
###############
# 1. remove the negative controls from the frequency table, 2. read the frequency table into phyloseq, 3. merge frequency table and metadata table, print phyloseq object
freqTable.noneg <- freqTable.lulu[, !names(freqTable.lulu) %in% negColumns]
OTU = otu_table(freqTable.noneg, taxa_are_rows = TRUE)
physeq.cor = merge_phyloseq(OTU, META)
physeq.cor
# 1. generate a dataframe with count data for ZOTU sequences, 2. add sample counts to the data frame, 3. plot using ggplot
readsumsdf = data.frame(nreads = sort(taxa_sums(physeq.cor), TRUE), sorted = 1:ntaxa(physeq.cor),
type = "ZOTUs")
readsumsdf = rbind(readsumsdf, data.frame(nreads = sort(sample_sums(physeq.cor),
TRUE), sorted = 1:nsamples(physeq.cor), type = "Samples"))
title = "Total number of reads"
p = ggplot(readsumsdf, aes(x = sorted, y = nreads)) + geom_bar(stat = "identity")
p + ggtitle(title) + facet_wrap(~type, 1, scales = "free")
p + ggtitle(title) + scale_y_log10() + facet_wrap(~type, 1, scales = "free")
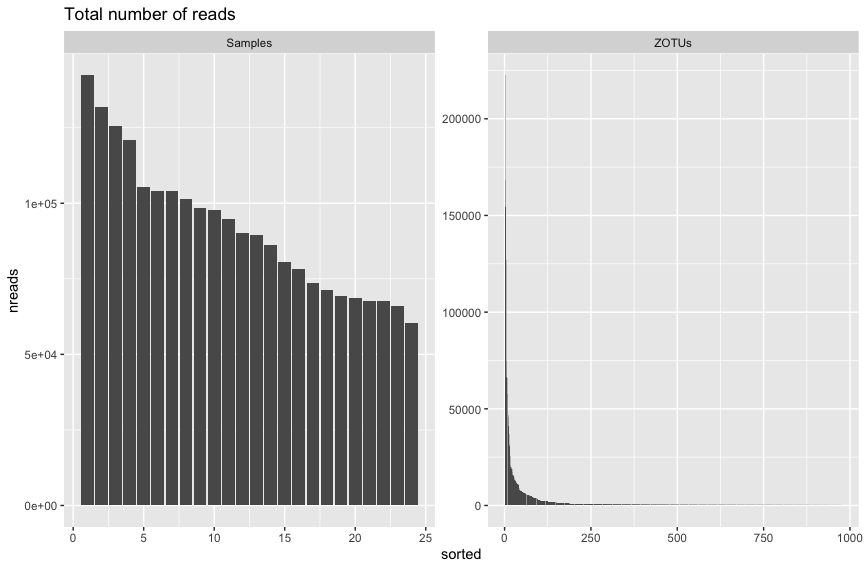
Fig. 34 : The read distribution for samples and ZOTU sequences without y-axis transformation.#
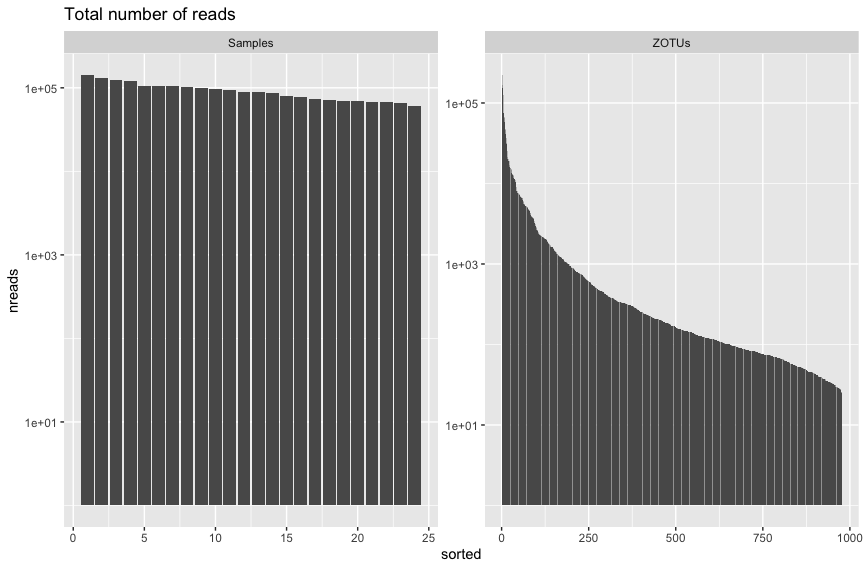
Fig. 35 : The read distribution for samples and ZOTU sequences with the y-axis log transformed.#
These figures above are fairly typical for metabarcoding data. There doesn’t seem to be too big a difference in the read count between samples. Additionally, the read distribution for ZOTU sequences looks pretty typical as well, whereby we have a few abundant sequences and a tail of sequences with low abundance.
7.2 Correlation#
To determine the correlation between total read count and ZOTU sequence richness, we can simply plot the total read count against the total number of observed ZOTU sequences within samples. In this tutorial, we run the spearman rank correlation over the pearson’s correlation, as the residuals are not normally distributed.
# 1. set total read count as vector, 2. set ZOTU richness as vector, 3. plot results, 4. draw linear regression
x <- as.vector(sample_sums(physeq.cor))
y <- as.vector(sample_sums(microbiome::transform(physeq.cor, 'pa')))
plot(x, y, xlab = 'sequencing depth', ylab = 'sequence richness',
main = 'sequence depth - richnes correlation', pch = 20, col = alpha('black', 0.4), cex = 3)
abline(lm(y ~ x), col = 'red')
# 1. create data frame with values, 2. run correlation test
df <- data.frame('Reads' = x, 'Richness' = y)
cor.test( ~ Reads + Richness, data = df, method = 'spearman', continuity = FALSE, conf.level = 0.95, exact = FALSE)
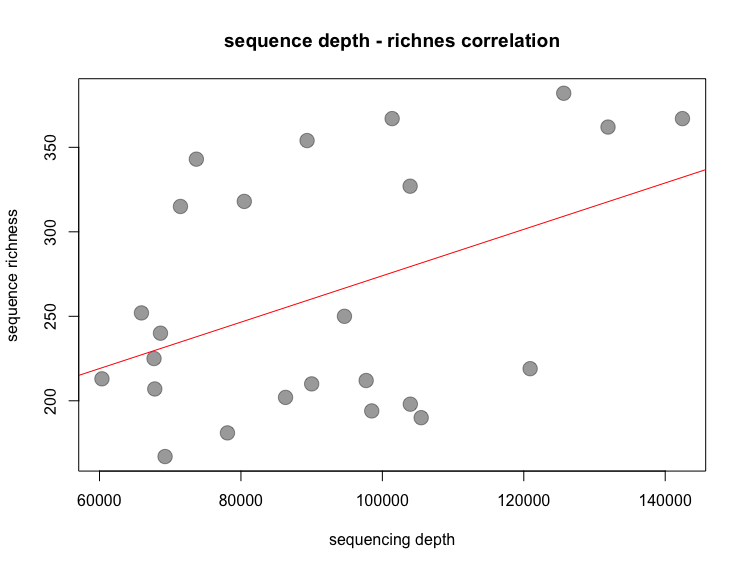
Fig. 36 : The correlation between read count and richness#
Output
Spearman's rank correlation rho
data: Reads and Richness
S = 1648.9, p-value = 0.1801
alternative hypothesis: true rho is not equal to 0
sample estimates:
rho
0.283105
While the p-value for the spearman rank correlation test is non-significant, a significant correlation was observed using pearson’s correlation. The latter, however, assumes a normal distribution of the residuals, which is violated in our case. Simulations on the violation have shown that pearson’s correlation is not too sensitive about this violation. As a conclusion, we can say that there is a weak correlation between sequencing depth and number of ZOTU sequences in a sample.
7.3 Rarefaction curves#
As a final check to see if we might want to rarefy our data, we can draw rarefaction curves and determine if they plateau. These graphs can also be used to determine if sufficient sequencing depth was achieved within our experiment. Such plots, however, need to be conducted on the unfiltered frequency table, as data processing has a major effect on the results of rarefaction curves and skews the data in favour of sufficient sequencing depth. These plots are mostly added to the supplementary files and referenced in the manuscript. We can use the function rarecurve from the vegan R package to draw rarefaction curves. We can also use the ampvis2 package to draw such curves and facet them based on metadata to provide a clearer picture.
########################
## RAREFACTION CURVES ##
########################
# read in the unfiltered data frames
freqTable.unfiltered <- read.table('zotutable.txt', header = TRUE, sep = '\t', row.names = 1, check.names = FALSE, comment.char = '')
metaData.unfiltered <- read.table('../0-metadata/metadata-COI-selected-updated.txt', header = TRUE, sep = '\t', row.names = 1, check.names = FALSE, comment.char = '')
taxonomyTable.unfiltered <- read.table('blastLineage.txt', header = TRUE, sep = '\t', row.names = 1, check.names = FALSE, comment.char = '')
# remove negative control samples, as they are not matching between data frames
negSamples <- grep('NTC', metaData.unfiltered$replicateLabel, value = TRUE)
freqTable.unfiltered <- freqTable.unfiltered[, !names(freqTable.unfiltered) %in% negSamples]
metaData.unfiltered <- metaData.unfiltered[!metaData.unfiltered$replicateLabel %in% negSamples, ]
# import files into phyloseq
OTU.unfiltered = otu_table(freqTable.unfiltered, taxa_are_rows = TRUE)
TAX.unfiltered = phyloseq::tax_table(as.matrix(taxonomyTable.unfiltered))
META.unfiltered = sample_data(metaData.unfiltered)
physeq.unfiltered = merge_phyloseq(OTU.unfiltered, TAX.unfiltered, META.unfiltered)
# run basic rarecurve function
rarecurve(as(t(otu_table(physeq.unfiltered)), 'matrix'), step = 100, xlab = "Sample Size", ylab = "Taxa")
# split rarefaction curves by group using the ampvis2 package
metadata.ampvis <- data.frame(sample_data(physeq.unfiltered), check.names = FALSE)
metadata.ampvis <- rownames_to_column(metadata.ampvis, var = "rowname")
asvTable.ampvis <- data.frame(otu_table(physeq.unfiltered), check.names = FALSE)
asvTable.ampvis$Species <- NA
ps3 <- amp_load(asvTable.ampvis, metadata.ampvis)
rarPlot <- amp_rarecurve(ps3, stepsize = 100, facet_by = 'island', color_by = 'SAMPLE') +
ylab('Number of observed ZOTUs')
rarPlot
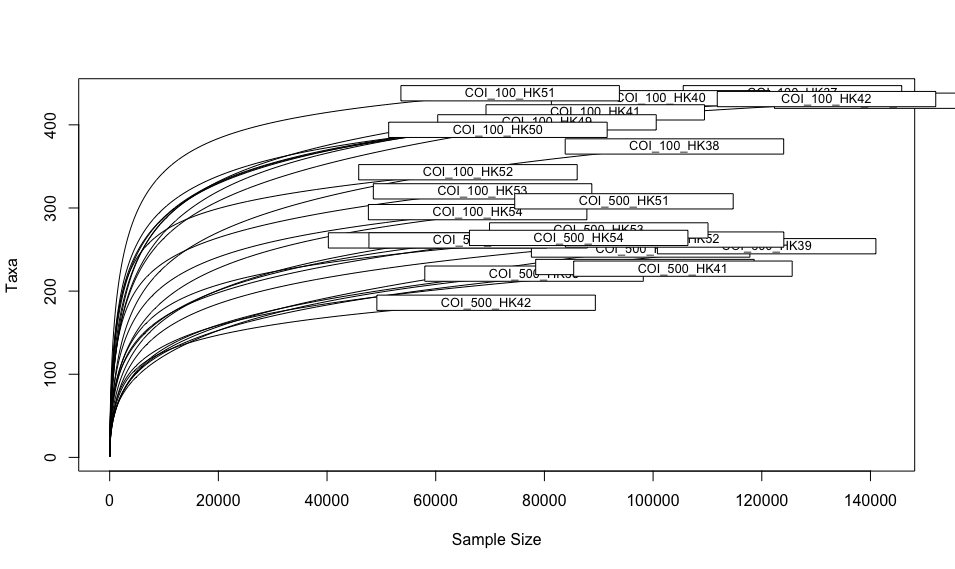
Fig. 37 : Rarefaction curves drawn with the rarecurve function in the vegan package.#
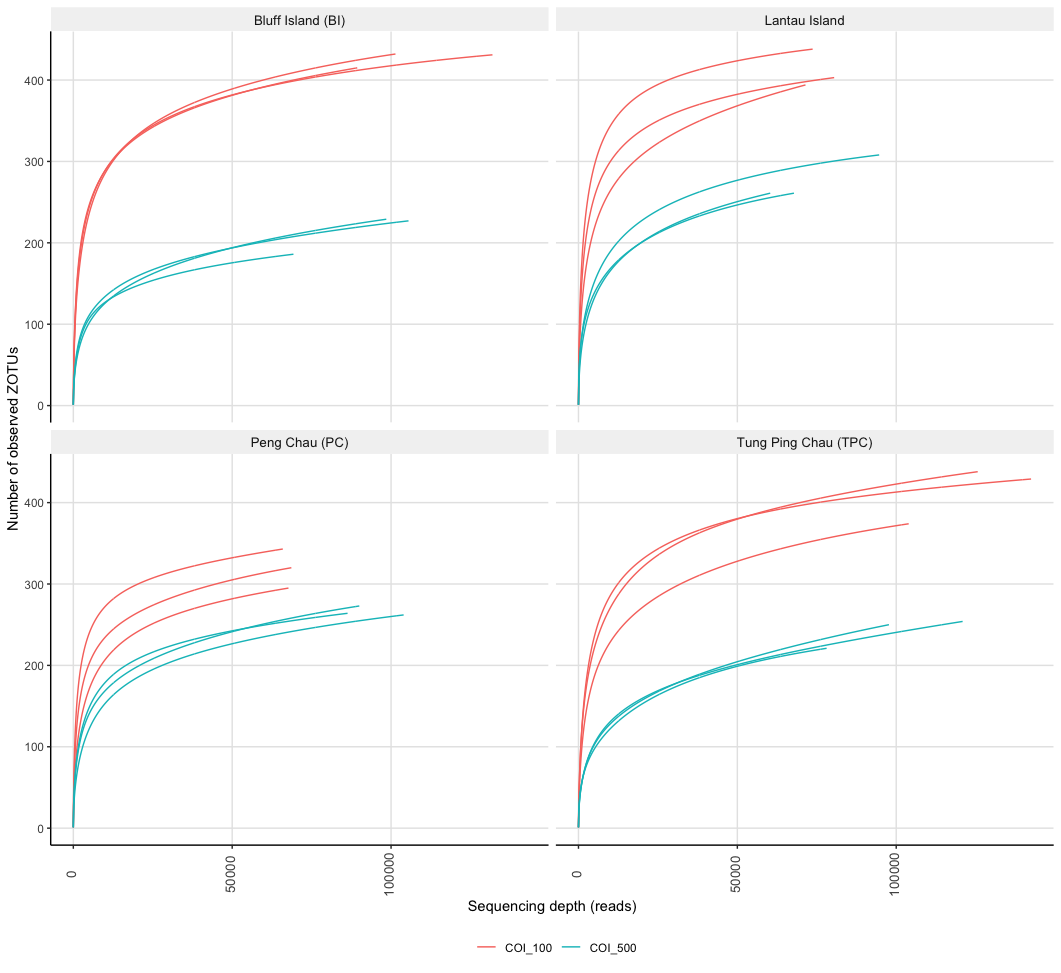
Fig. 38 : Rarefaction curves drawn with the amp_rarecurve function in the ampvis2 package. Rarefaction curves facetted by location and coloured by size fraction.#
8. Tutorial data#
Now that we have covered several considerations with regards to pre-processing the data prior to the statistical analysis, let’s decide how we will process the tutorial data set before moving on to the next section. For the tutorial data set, we will use a combined approach to deal with detections in negative controls, whereby we first run the decontam package and for sequences not identified as contaminants, but a positive detection in the negative controls, we will set an abundance threshold of 10x the read count in the negative controls to keep a “true” detection. We will set an abundance threshold whereby we remove singleton detections, due to the reads found in the negative control samples. Next, we will remove artefact sequences through the LULU algorithm. Last, we will remove ZOTU sequences for which no BLAST hit could be obtained. Finally, we will update and export the filtered dataframes to be used in the next sessions.
#########################
# PREPARE R ENVIRONMENT #
#########################
library(Biostrings)
library(dplyr)
library(decontam)
library(phyloseq)
library(ggplot2)
library(lulu)
library(microbiome)
library(scales)
library(vegan)
library(ampvis2)
library(tidyverse)
# set working directory
setwd('/Users/gjeunen/Documents/work/lectures/HKU2023/tutorial/sequenceData/8-final')
##################
# READ DATA IN R #
##################
metaData <- read.table('../0-metadata/metadata-COI-selected-updated.txt', header = TRUE, sep = '\t', row.names = 1, check.names = FALSE, comment.char = '')
freqTable <- read.table('zotutable.txt', header = TRUE, sep = '\t', row.names = 1, check.names = FALSE, comment.char = '')
taxonomyTable <- read.table('blastLineage.txt', header = TRUE, sep = '\t', row.names = 1, check.names = FALSE, comment.char = '')
sequenceTable <- readDNAStringSet('zotus.fasta')
#####################
# NEGATIVE CONTROLS #
#####################
# 1. import the frequency table into phyloseq, 2. import the metadata table into phyloseq, 3. merge phyloseq objects
OTU = otu_table(freqTable, taxa_are_rows = TRUE)
META = sample_data(metaData)
physeq = merge_phyloseq(OTU, META)
physeq
# 1. create new column with logical variables, 2. determine contaminants, 3. return summary table, 4. display first entries
sample_data(physeq)$is.neg <- sample_data(physeq)$island == 'control'
contamdf.prev <- isContaminant(physeq, method = 'prevalence', neg = 'is.neg', threshold = 0.5)
table(contamdf.prev$contaminant)
head(which(contamdf.prev$contaminant))
# 1. identify contaminants, 2. remove contaminants from frequence table, 3. remove contaminants from sequence list
contamSeq <- row.names(contamdf.prev)[with(contamdf.prev, contaminant %in% c(TRUE))]
freqTable.clean <- freqTable[!row.names(freqTable) %in% contamSeq, ]
sequenceTable.clean <- sequenceTable[rownames(freqTable.clean)]
# 1. find columns that contain NTC in header, 2. sum rows that contain NEG in header to column NEGSUM, 3. drop individual NEG columns
negColumns <- grep('NTC', names(freqTable.clean), value = TRUE)
freqTable.clean$NEGSUM <- rowSums(freqTable.clean[negColumns])
freqTable.clean <- freqTable.clean[, !names(freqTable.clean) %in% negColumns]
# 1. initialise new df, 2. for loop to set all values less than 10 x the negative control value to 0
freqTable.clean.relaxedFilter <- freqTable.clean
negZOTUs <- rownames(freqTable.clean.relaxedFilter)[which(freqTable.clean.relaxedFilter$NEGSUM > 0)]
for (negZOTU in negZOTUs) {
condition <- freqTable.clean.relaxedFilter[negZOTU, 'NEGSUM'] * 10
freqTable.clean.relaxedFilter[negZOTU, -1] <- ifelse(freqTable.clean.relaxedFilter[negZOTU, -1] > condition, freqTable.clean.relaxedFilter[negZOTU, -1], 0)
}
#############
# ABUNDANCE #
#############
# 1. set all values lower than 2 to 0, 2. check if lowest value in dataframe is 2, excluding 0
freqTable.clean.relaxedFilter[freqTable.clean.relaxedFilter < 2] <- 0
min(freqTable.clean.relaxedFilter[freqTable.clean.relaxedFilter != 0], na.rm = TRUE)
# 1. remove columns that sum to 0, 2. remove rows that sum to 0
colsRemove <- colSums(freqTable.clean.relaxedFilter) == 0
freqTable.clean.relaxedFilter <- freqTable.clean.relaxedFilter[, !colsRemove]
rowsRemove <- rowSums(freqTable.clean.relaxedFilter) == 0
freqTable.clean.relaxedFilter <- freqTable.clean.relaxedFilter[!rowsRemove, ]
# 1. update dataframes to match names, 2. export sequence file
sequenceTable.clean.relaxedFilter <- sequenceTable.clean[rownames(freqTable.clean.relaxedFilter)]
taxonomyTable.clean.relaxedFilter <- taxonomyTable[rownames(freqTable.clean.relaxedFilter), ]
metaData.clean.relaxedFilter <- metaData[colnames(freqTable.clean.relaxedFilter), ]
writeXStringSet(sequenceTable.clean.relaxedFilter, file = 'zotusInterim.fasta')
cd /Users/gjeunen/Documents/work/lectures/HKU2023/tutorial/sequenceData/8-final
makeblastdb -in zotusInterim.fasta -parse_seqids -dbtype nucl
blastn -db zotusInterim.fasta -outfmt '6 qseqid sseqid pident' -out match_list.txt -qcov_hsp_perc 80 -perc_identity 84 -query zotusInterim.fasta
########
# LULU #
########
# 1. read in the BLAST results, 2. run the lulu algorithm, 3. determine the number of artefact sequences, 4. export new frequency table from LULU
matchlist <- read.table('match_list.txt', header = FALSE, as.is = TRUE, stringsAsFactors = FALSE)
curatedResult <- lulu(freqTable.clean.relaxedFilter, matchlist)
curatedResult$discarded_count
freqTable.clean.relaxedFilter.lulu <- curatedResult$curated_table
# 1. update tables
sequenceTable.clean.relaxedFilter.lulu <- sequenceTable.clean.relaxedFilter[rownames(freqTable.clean.relaxedFilter.lulu)]
taxonomyTable.clean.relaxedFilter.lulu <- taxonomyTable.clean.relaxedFilter[rownames(freqTable.clean.relaxedFilter.lulu), ]
metaData.clean.relaxedFilter.lulu <- metaData.clean.relaxedFilter[colnames(freqTable.clean.relaxedFilter.lulu), ]
# 1. remove ZOTUs without a BLAST hit
lowConfidenceTax <- rownames(taxonomyTable.clean.relaxedFilter.lulu)[which(is.na(taxonomyTable.clean.relaxedFilter.lulu$order))]
freqTable.clean.relaxedFilter.lulu.taxFilter <- freqTable.clean.relaxedFilter.lulu[!(rownames(freqTable.clean.relaxedFilter.lulu) %in% lowConfidenceTax), ]
# 1. update tables
sequenceTable.clean.relaxedFilter.lulu.taxFilter <- sequenceTable.clean.relaxedFilter.lulu[rownames(freqTable.clean.relaxedFilter.lulu.taxFilter)]
taxonomyTable.clean.relaxedFilter.lulu.taxFilter <- taxonomyTable.clean.relaxedFilter.lulu[rownames(freqTable.clean.relaxedFilter.lulu.taxFilter), ]
metaData.clean.relaxedFilter.lulu.taxFilter <- metaData.clean.relaxedFilter.lulu[colnames(freqTable.clean.relaxedFilter.lulu.taxFilter), ]
# 1. export tables
write.table(freqTable.clean.relaxedFilter.lulu.taxFilter, 'zotutableFiltered.txt', append = FALSE, sep = '\t', dec = '.', row.names = TRUE, col.names = NA)
write.table(metaData.clean.relaxedFilter.lulu.taxFilter, '../0-metadata/sampleMetadataFiltered.txt', append = FALSE, sep = '\t', dec = '.', row.names = TRUE)
write.table(taxonomyTable.clean.relaxedFilter.lulu.taxFilter, 'taxonomyFiltered.txt', append = FALSE, sep = '\t', dec = '.', row.names = TRUE, col.names = NA)
writeXStringSet(sequenceTable.clean.relaxedFilter.lulu.taxFilter, file = 'zotusFiltered.fasta')
That is it for this section! We hope you have gained a better understanding of some of the pre-processing and “data massaging” that is undertaken in metabarcoding research projects.